血液蛋白质组学专题
Blood proteomics route map
血液蛋白质组学研究技术路线和难点
血液样本常用于生物标志物研究中的早筛假设、分层、预后和监测探索,但动态范围、预分析变量、高丰度蛋白、细胞污染和平台间读数差异会同时影响结果解释。专题把亲和 Panel、开放 MS、选择性富集 MS、PRM 验证、DeePB 前处理和 QC 体系放在同一条可学习路径中。
Olink / SomaScan / NULISA
DIA-MS / PRM
DeePB 富集
血液 QC
项目怎么判断
血液项目先回答三个问题
先判断项目是做发现、验证还是研究评估;再选择 Panel、开放 MS 或富集 MS;最后确认样本质量、前处理记录和数据 QC 是否足以支持结果解释。
项目阶段
项目现在是发现、验证还是评估?
血液蛋白质组学常用于早筛假设、分层、预后和监测探索。平台选择不能只看检测数量,而要先明确项目处于发现、收敛、验证还是研究评估阶段。
- Panel 平台用于快速扩大候选空间,适合已知通路和大队列筛查。
- 开放 MS 和富集 MS 用于补充 Panel 外信息,并保留肽段和谱图证据。
- 候选收敛阶段需要同时看统计信号、机制解释、临床变量和批次稳定性。
- 验证评估阶段应引入 PRM/PQ500、ELISA 或其他正交方法,确认候选物读数是否稳定。
Panel 筛选
Olink、SomaScan、NULISA 等平台用于高通量候选筛查。
开放发现
DIA-MS、修饰、突变、肽组和富集 MS 提供肽段证据链。
候选收敛
把统计信号、机制解释和临床变量整合后筛掉不稳定候选。
验证确认
PRM、ELISA 或其他正交方法确认候选物的可重复读数。
研究评估
进入独立队列、临床变量、阈值探索和报告解释规则评估。
资料拆解
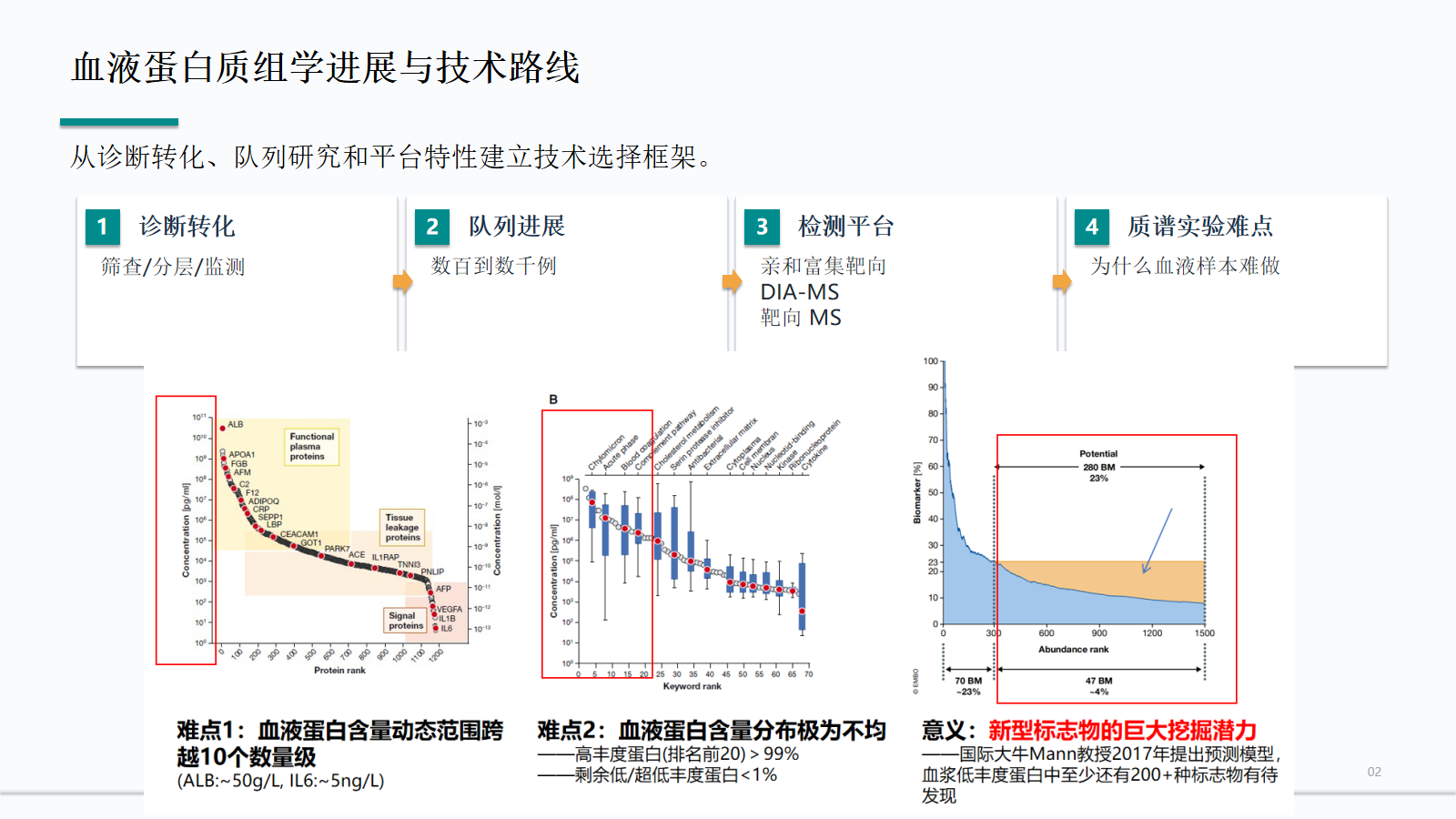
血液蛋白质组学进展与技术路线
血液蛋白质组学首先服务生物标志物研究中的筛查假设、分层、预后和监测问题。路线选择必须同时考虑队列规模、平台读数类型和质谱实验难点。
- 从诊断转化、队列研究和平台特性建立技术选择框架。
查看原图
资料拆解
Biomarker 发现路线
biomarker 项目建议按发现、收敛、验证和研究评估分层推进。发现阶段追求候选空间,验证阶段追求稳定读数,研究评估阶段强调独立队列和可解释路径。
- 发现阶段要广,验证阶段要准,转化阶段要能解释、能复现、能进入临床研究路径。
查看原图

资料拆解
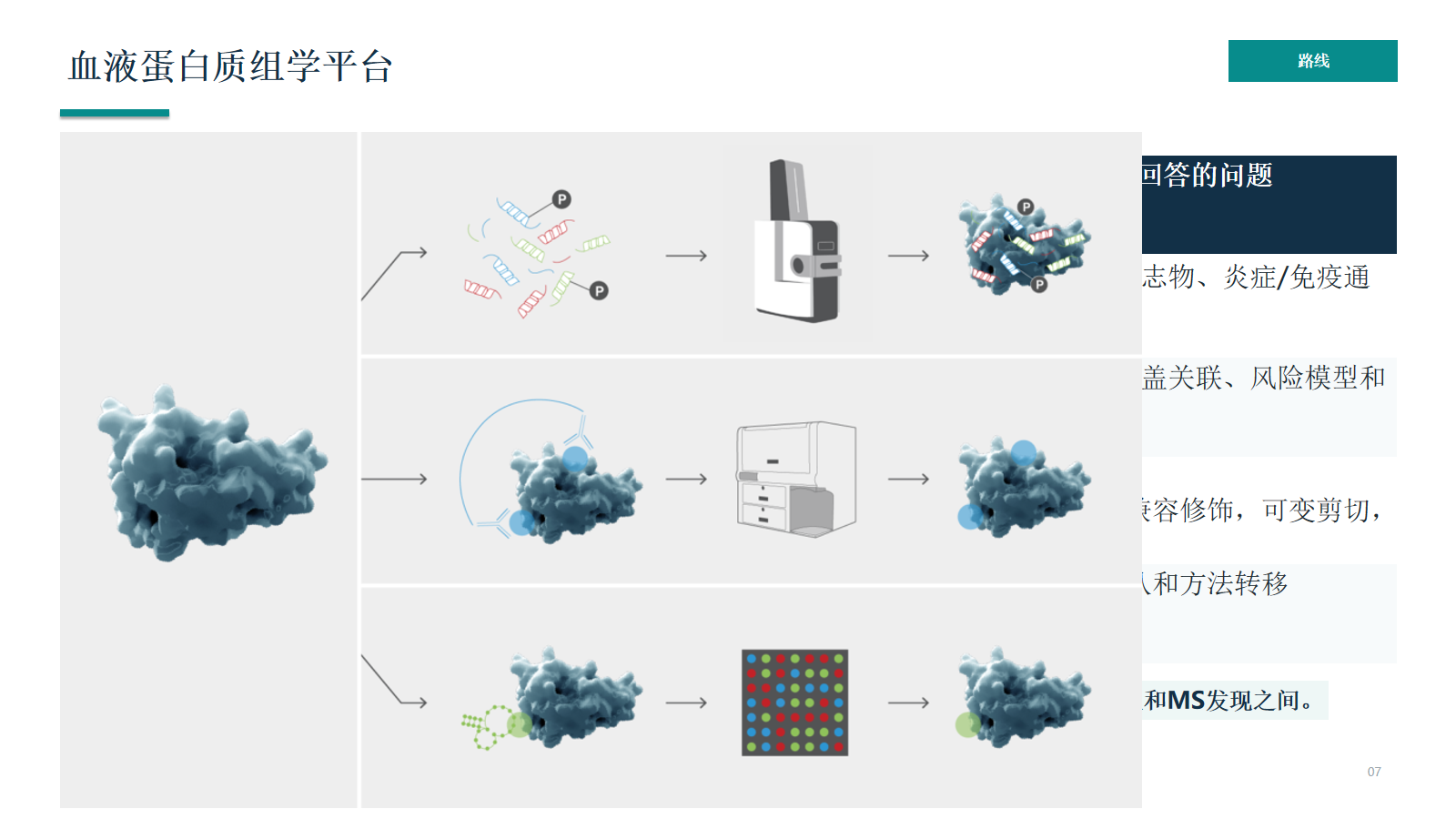
血液蛋白质组学平台
亲和平台、MS discovery 和 Seer/蛋白冠类路线分别处于不同位置:亲和平台更偏 Panel 内发现,MS 更偏肽段证据和正交验证,蛋白冠路线属于样本前处理与 MS discovery 的中间层。
- 核心原则:亲和平台适合“Panel内发现”,MS适合“肽段证据和正交验证”;Seer介于样本前处理和MS发现之间。
查看原图
平台选择
该用 Panel、开放 MS 还是富集 MS?
Olink、SomaScan 和 NULISA 通过抗体或适配体获得高通量读数;纳米材料/蛋白冠富集改变血浆可观测空间;MS 提供肽段证据链。它们应该组合使用,而不是互相替代。
- Olink/PEA 适合低样本量、大队列、免疫炎症相关 Panel 内发现。
- SomaScan 覆盖广、缺失少,但需要把适配体结合信号和蛋白真实浓度区分开。
- NULISA 更接近高灵敏多重免疫检测,适合低丰度免疫标志物的稳定读数。
- 纳米磁珠/蛋白冠路线属于选择性采样,应重点评估材料批次、污染和回收稳定性。
- 平台比较应按发现覆盖、队列通量、肽段证据、定量收敛和主要风险分维度判断。
资料拆解
Olink / PEA:双抗体邻近延伸的大队列平台
Olink/PEA 的优势来自双抗体邻近延伸和核酸读出。它适合低样本量、高通量和低丰度免疫/炎症蛋白筛查,但结果仍受 Panel、表位、批次和 NPX 相对定量限制。
- 优势来自双抗体识别和NGS读出;限制来自Panel内靶点和表位依赖。
- 适合批量样本
- 适用定位:大样本Panel内发现和遗传流行病学;关键候选物仍需MS/ELISA/PRM等正交验证。
查看原图

资料拆解
SomaLogic / SomaScan:SOMAmer适配体高通量测量
SomaScan 以 SOMAmer 适配体结合信号进行高通量测量,覆盖范围大、缺失值通常较少。解释时要记住读数来自适配体结合,不等同于所有蛋白型的真实浓度。
- 覆盖面广、低缺失值友好;但单适配体结合信号需要谨慎。
- 保留结合复合物
- 需要跨版本校准
- 适合广覆盖关联和风险模型。
- 适合探索性大Panel筛选。
查看原图

资料拆解
NULISA:高灵敏免疫-核酸读出的低丰度Panel
NULISA 更像高灵敏多重免疫检测,适合低丰度炎症/免疫蛋白、PK/PD、毒理和纵向队列。它的强项是灵敏度和部分绝对定量,边界是 Panel 化和表位依赖。
- 更像高灵敏、多重免疫检测,强项是低丰度炎症/免疫蛋白的稳定测量。
- 部分目标提供AQ
- 用于纵向队列
- AQ版本对150+目标提供绝对定量。
- CV表现好;适合细胞因子、
查看原图

资料拆解
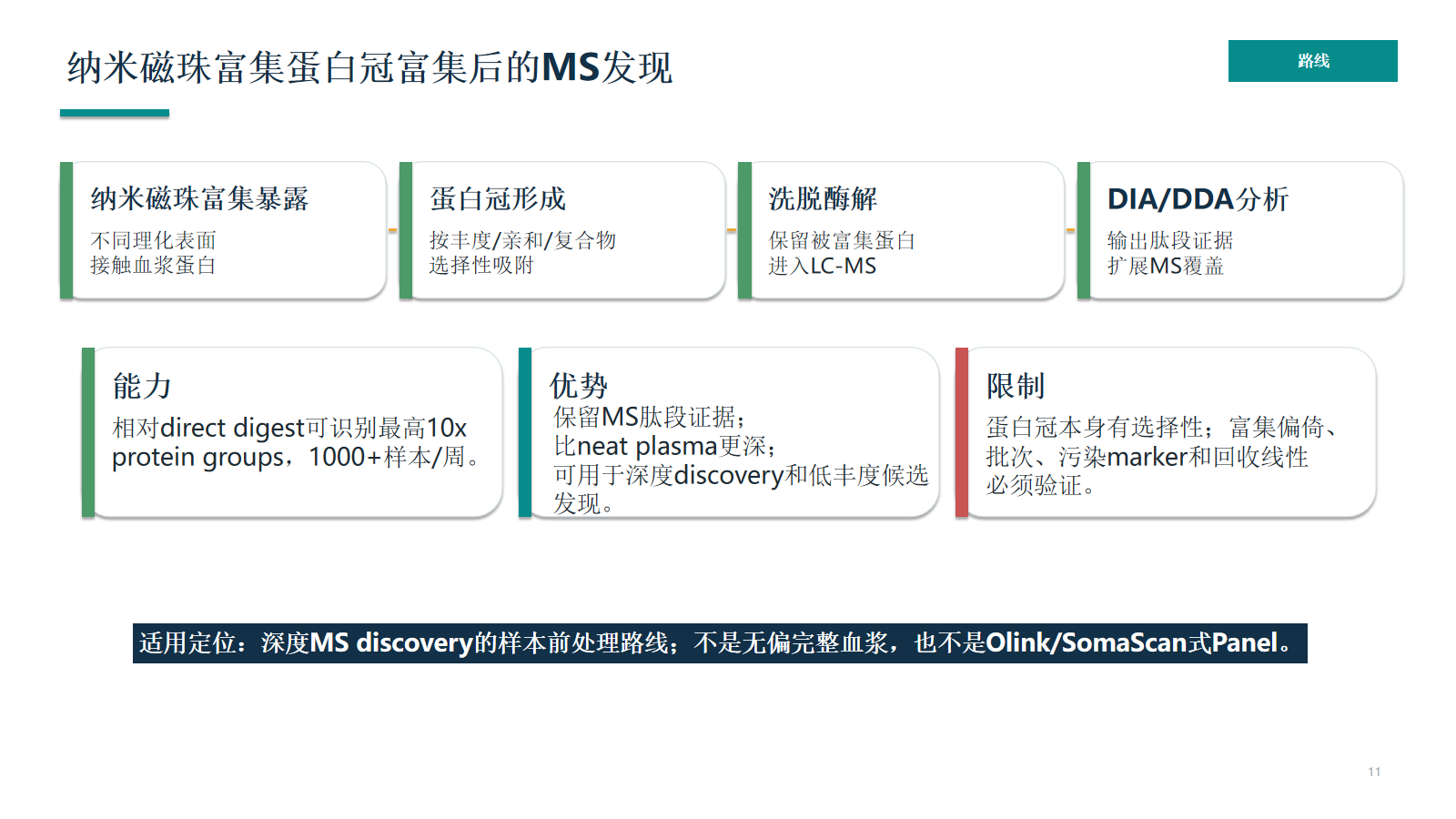
纳米磁珠富集蛋白冠富集后的MS发现
纳米磁珠/蛋白冠富集把血浆蛋白按理化表面和亲和关系进行选择性采样,从而改变可观测蛋白空间。它能加深 MS discovery,但必须验证选择性、批次和污染风险。
- 保留被富集蛋白
- 进入LC-MS
- protein groups,1000+样本/周。
- 保留MS肽段证据;
- 可用于深度discovery和低丰度候选
查看原图
资料拆解
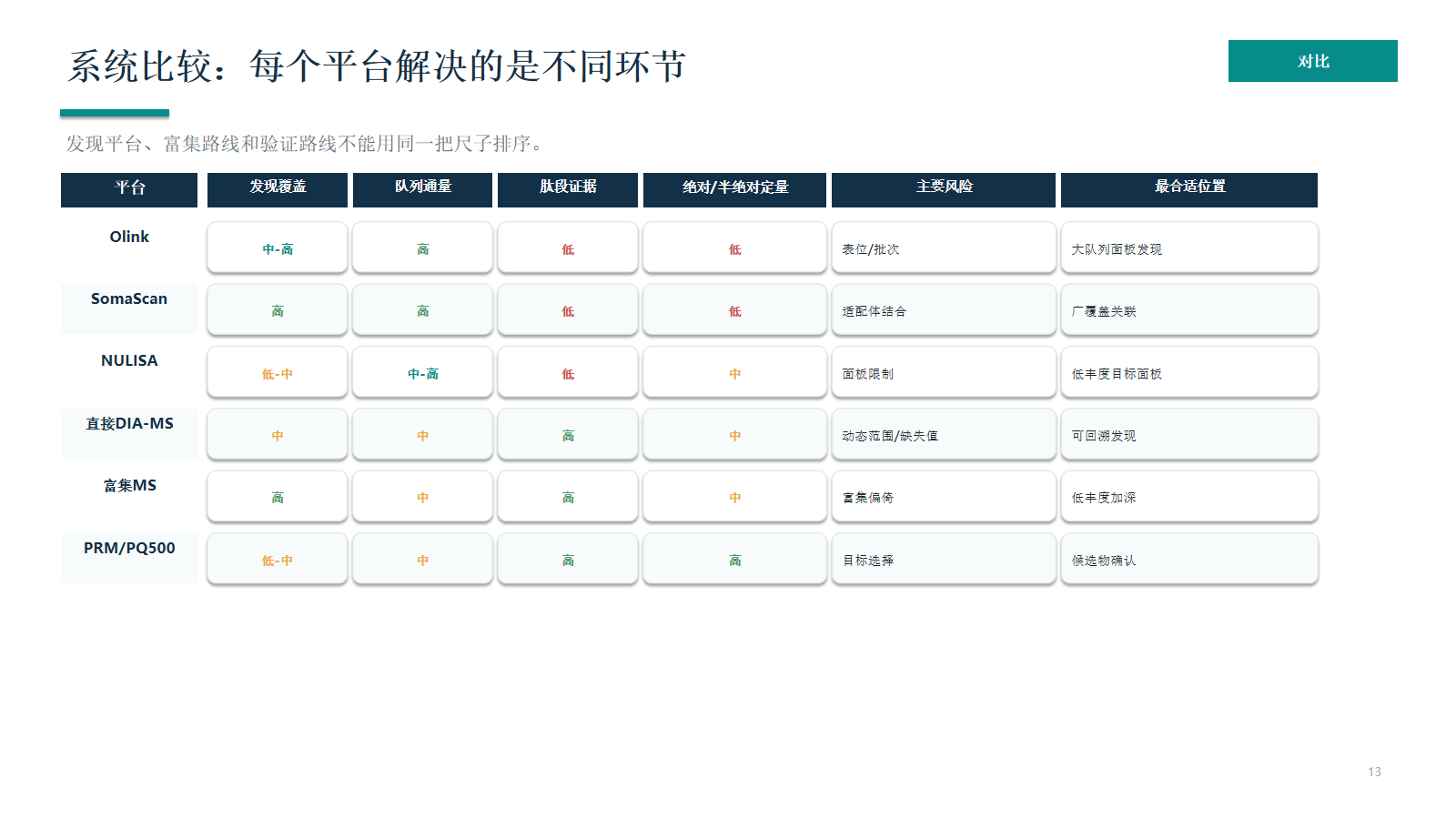
系统比较:每个平台解决的是不同环节
不同平台要放在同一项目链条里比较:Olink/SomaScan/NULISA 强在 Panel 或低丰度免疫读数,DIA-MS 强在可回溯发现,富集 MS 强在扩展可观测空间,PRM/PQ500 强在候选确认。
- 发现平台、富集路线和验证路线不能用同一把尺子排序。
- 候选物确认
查看原图
资料拆解
亲和平台和 MS 的边界
亲和 Panel 和 MS 不是互相替代关系。亲和平台适合万人队列和已知通路筛选;MS 适合 Panel 外 discovery、PTM/蛋白型和机制解释,但对动态范围和批次效应更敏感。
- 优势:通量高、样本量低、适合万人队列
- 适合:风险模型、pQTL、已知通路筛选
- 适合:开放 discovery、PTM/蛋白型、
查看原图

结果复核
质谱结果能不能回到肽段和谱图?
血液 MS 不应只追求蛋白数量。真正决定结果能否解释的是谱图、肽段、蛋白型、修饰、缺失值控制和候选物验证路径是否完整。
- 开放 discovery 负责发现 Panel 外蛋白、修饰和机制线索。
- 亲和读数和 MS 覆盖集合不同,联合分析时要分清共同候选与平台特异候选。
- ApuQuant 等算法可以改善峰识别和缺失值控制,但不能替代批次 QC、峰形复核和独立验证。
- PRM/MRM/PQ500 适合把 discovery 候选推进到半绝对或绝对定量验证。
资料拆解
质谱路线:从开放发现到候选物确认
MS 路线的核心价值不是把蛋白数做得最大,而是保留肽段证据链,使结果可以回到谱图、肽段、蛋白型、PTM 或候选物验证层面复核。
- MS的核心价值是肽段证据链,而不是单纯追求蛋白数量。
查看原图

资料拆解
不同技术路线的定位
从通量和蛋白证据两个维度看,亲和平台通常通量高但缺少肽段证据;MS 和富集 MS 证据链更完整,但需要更强的 QC 和批次管理。
- 从通量和蛋白证据两个维度看,亲和平台通常通量高但缺少肽段证据;MS 和富集 MS 证据链更完整,但需要更强的 QC 和批次管理。
查看原图

资料拆解
不同技术路线的定位
如果问题涉及肽段、修饰或蛋白型,MS 路线的优势会更明显。亲和读数可以给出候选信号,但通常不能直接回答具体肽段或修饰位点问题。
- 如果问题涉及肽段、修饰或蛋白型,MS 路线的优势会更明显。亲和读数可以给出候选信号,但通常不能直接回答具体肽段或修饰位点问题。
查看原图
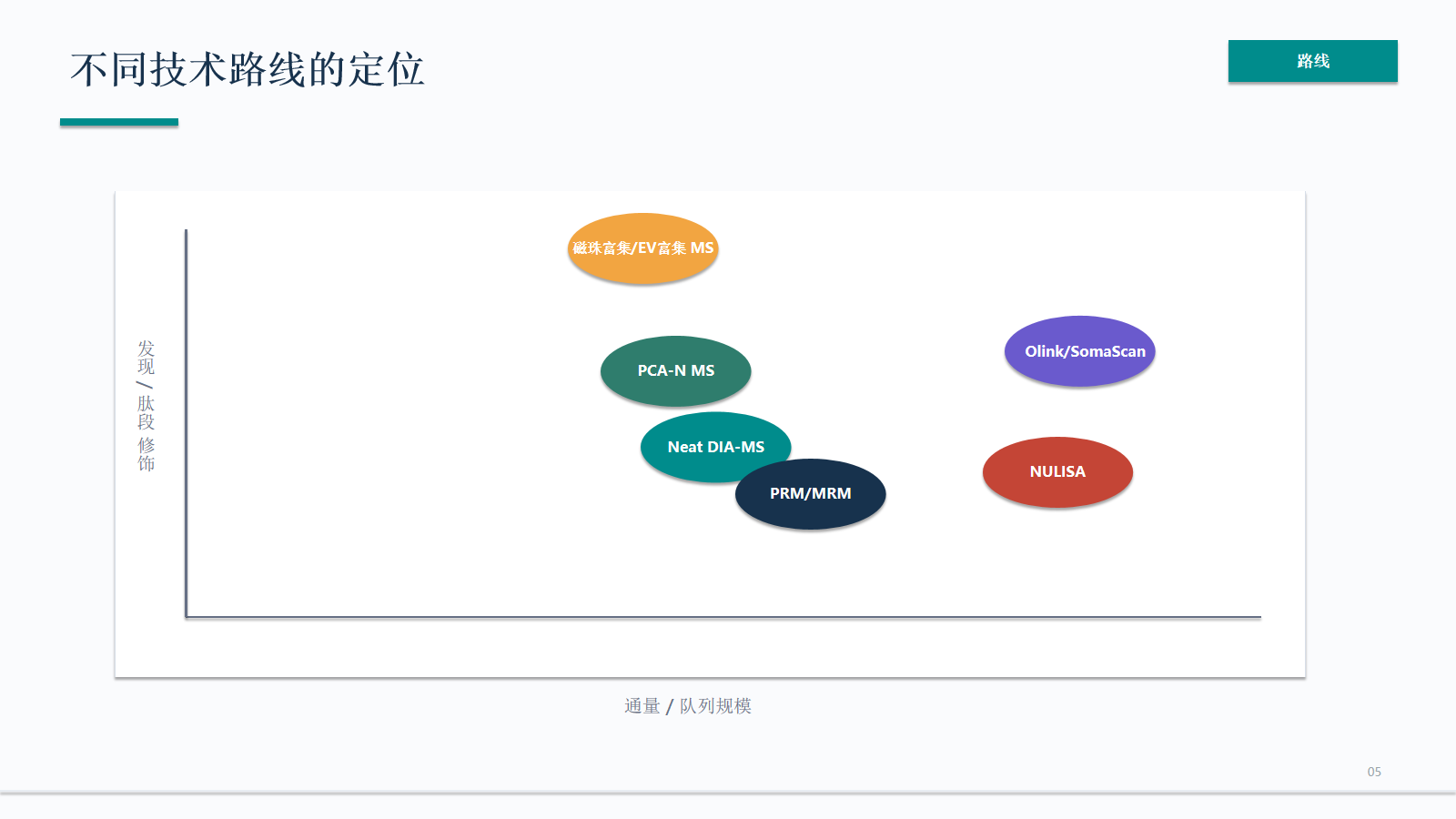
资料拆解
不同技术路线的定位
检测灵敏度维度不能单独决定路线。NULISA、Olink/SomaScan、富集 MS 和 PRM 都可能覆盖低丰度目标,但它们的读数机制、可回溯性和验证成本不同。
- 检测灵敏度维度不能单独决定路线。NULISA、Olink/SomaScan、富集 MS 和 PRM 都可能覆盖低丰度目标,但它们的读数机制、可回溯性和验证成本不同。
查看原图
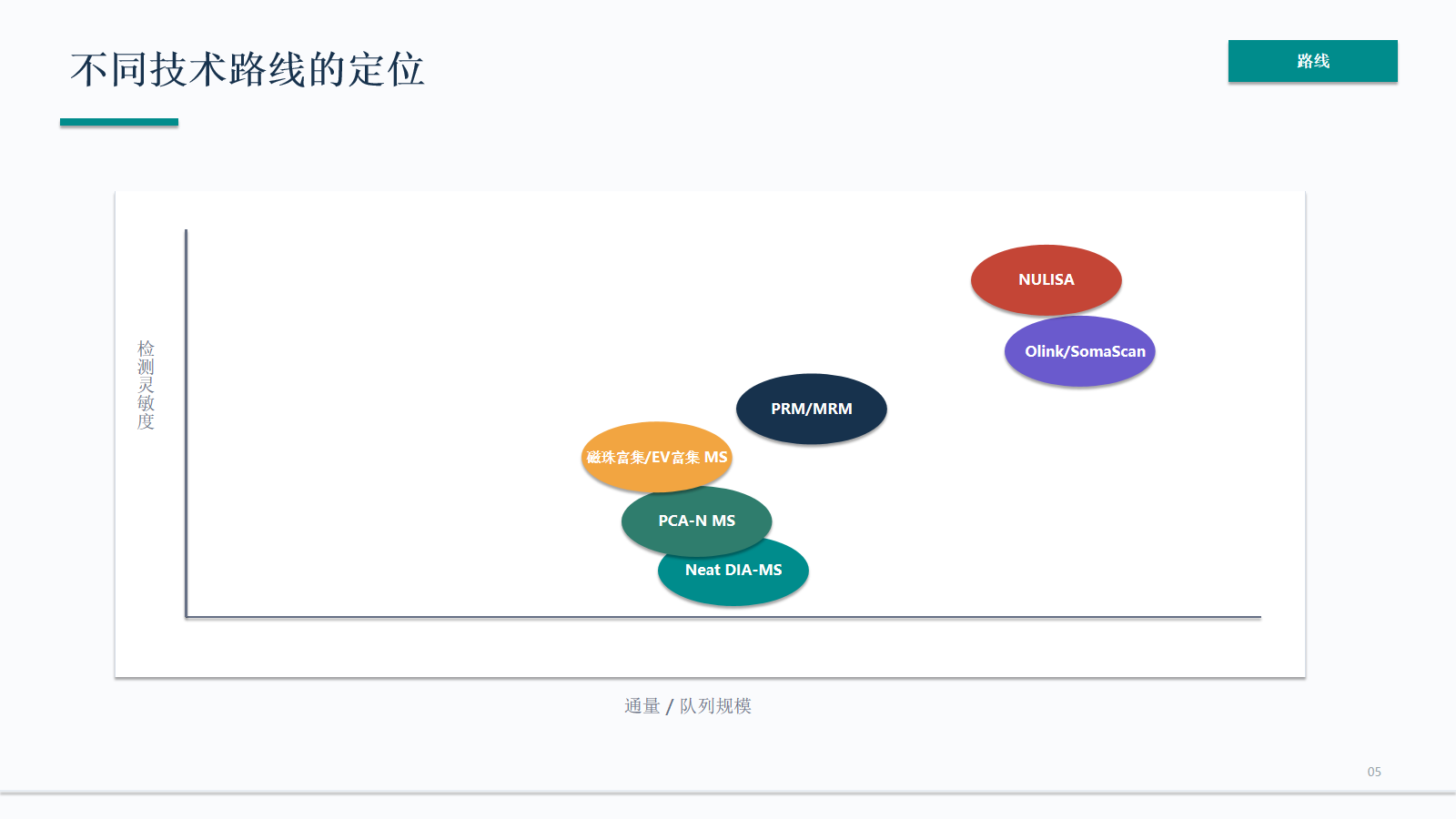
资料拆解
误差来源:每个平台都需要自己的QC
每个平台都有自己的误差来源。亲和平台要关注表位、适配体和批次;MS 要关注动态范围、缺失值和谱图复杂度;富集路线要关注选择性采样和污染。
- 亲和平台重点检查表位、适配体、批次和跨 panel 一致性。
- MS 路线重点检查动态范围、缺失值、谱图复杂度和采集稳定性。
- 富集路线重点检查选择性采样、污染 marker、回收率和材料批次。
查看原图

资料拆解
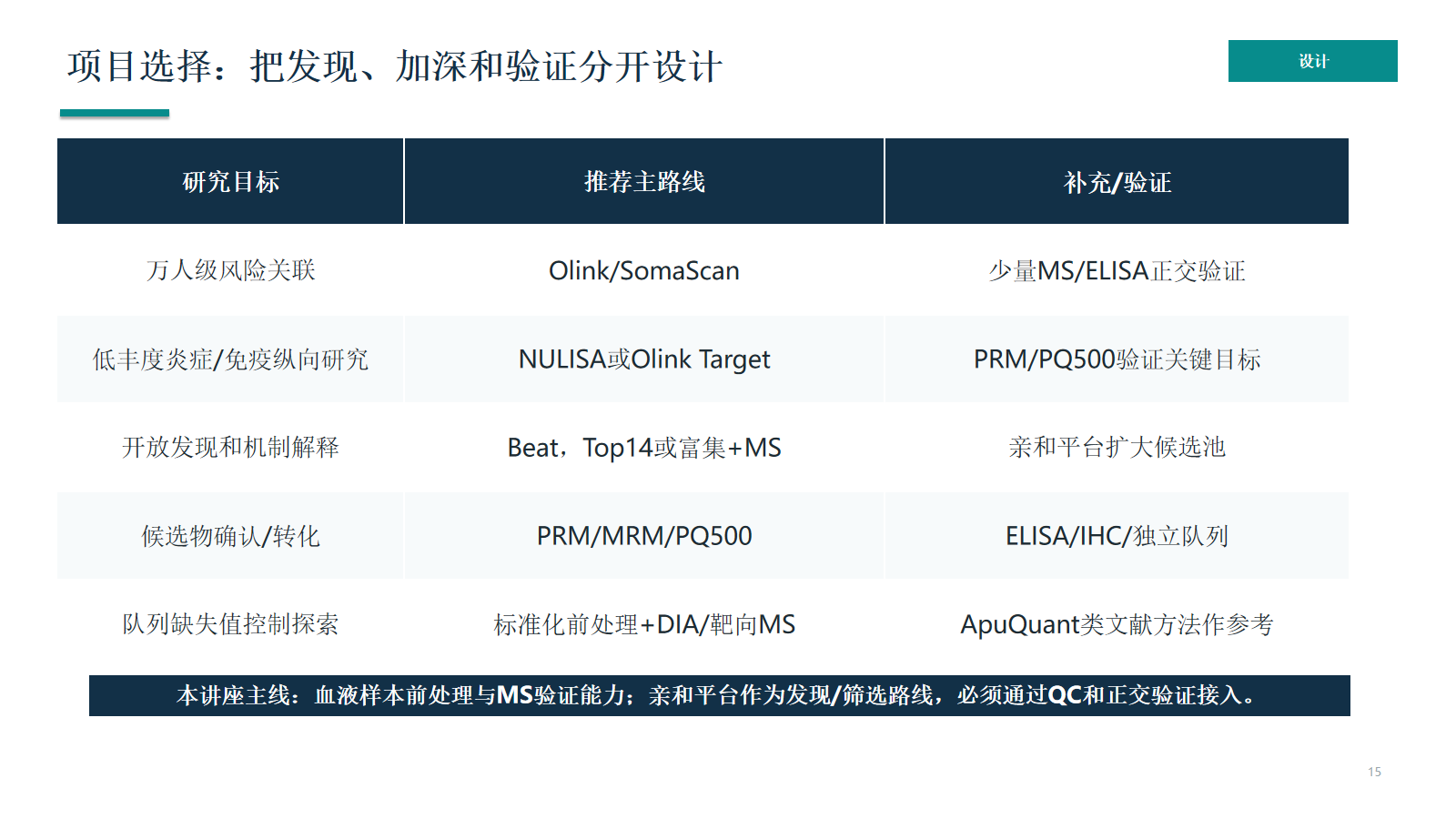
项目选择:把发现、加深和验证分开设计
项目设计应把发现、加深和验证分开。亲和平台可以提供发现/筛选信号,MS 提供肽段证据,PRM/ELISA 等正交方法负责候选物确认。
- 研究判断顺序:血液样本前处理与MS验证能力;亲和平台作为发现/筛选路线,必须通过QC和正交验证接入。
查看原图
资料拆解
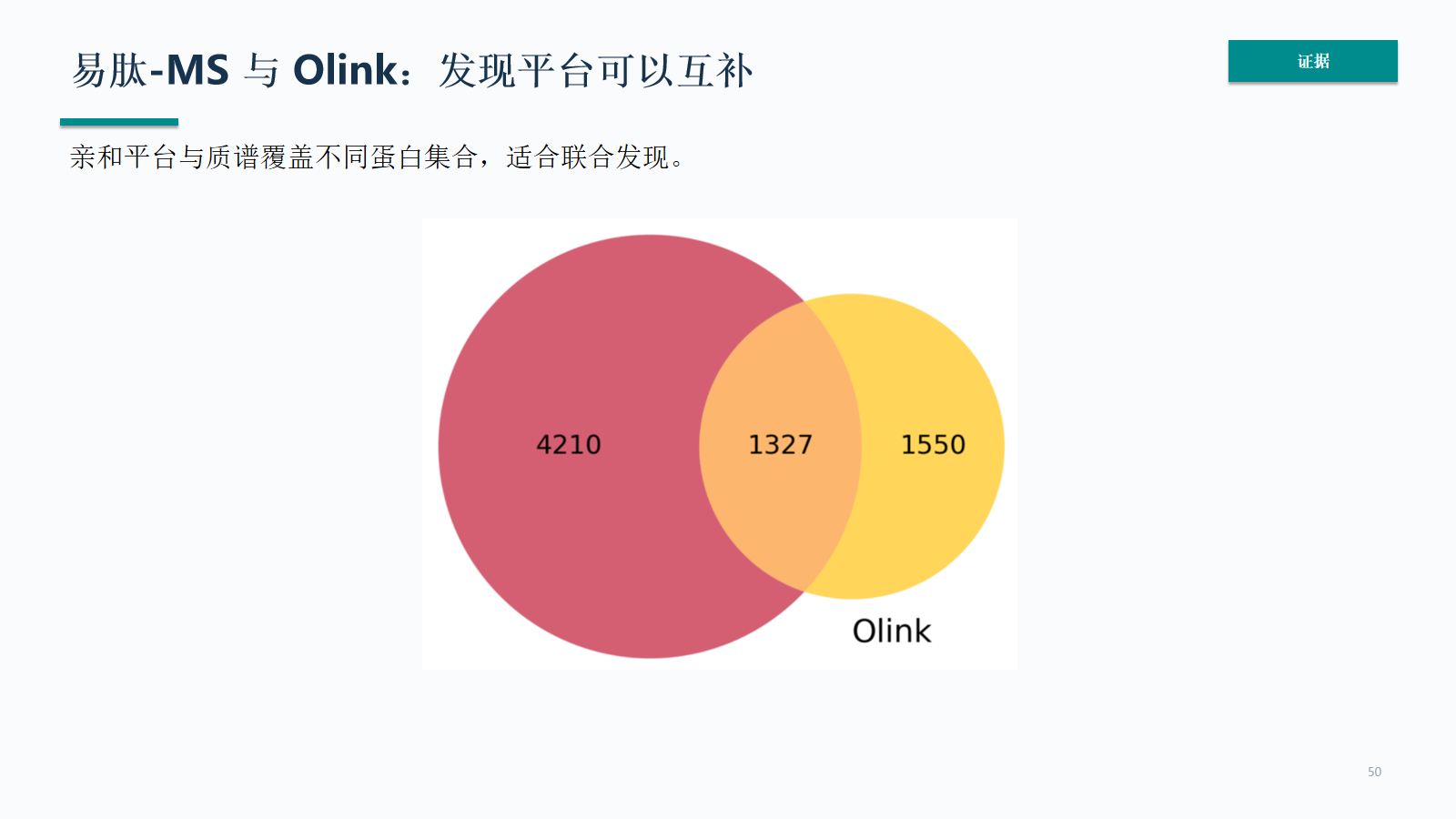
易肽-MS 与 Olink:发现平台可以互补
易肽-MS 与 Olink 的互补性来自覆盖集合不同。联合分析时要关注两类平台共同出现的候选、仅在某一平台出现的候选,以及这些差异是否符合生物学和技术机制。
- 亲和平台与质谱覆盖不同蛋白集合,适合联合发现。
查看原图
资料拆解
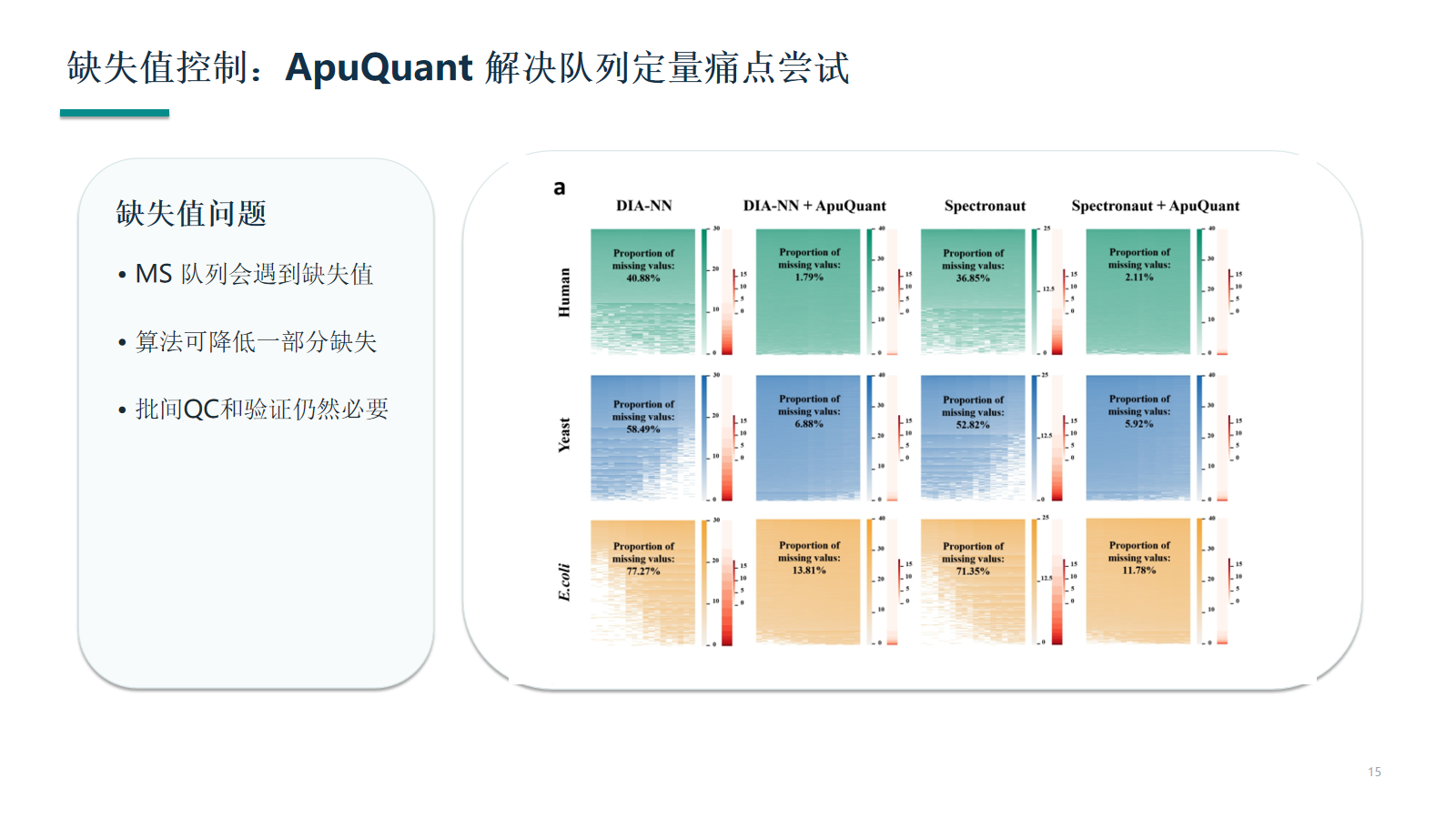
缺失值控制:ApuQuant 解决队列定量痛点尝试
MS 队列定量会遇到缺失值。ApuQuant 等算法可以降低一部分缺失,但算法处理不能替代批间 QC、原始峰形复核和独立验证。
- 缺失值要先判断是低丰度边界、批次漂移、峰识别错误还是样本质量问题。
- 算法可辅助降低缺失,但不能替代 pooled QC、峰形复核和独立验证。
- 缺失集中在某批次或某分组时,应优先排查技术来源。
查看原图
资料拆解
缺失值控制:ApuQuant 解决队列定量痛点尝试
缺失值控制需要同时看算法、采集质量和样本批次。若缺失集中在某一批次、某一类样本或某一低丰度区域,应优先判断技术来源。
- 缺失率分布需要按批次、分组、丰度区间和样本类型分层查看。
- 候选物进入验证前,应确认缺失不是由峰形质量或采集窗口造成。
- ApuQuant 类算法适合作为一致性改善工具,不适合作为质量问题掩盖工具。
查看原图
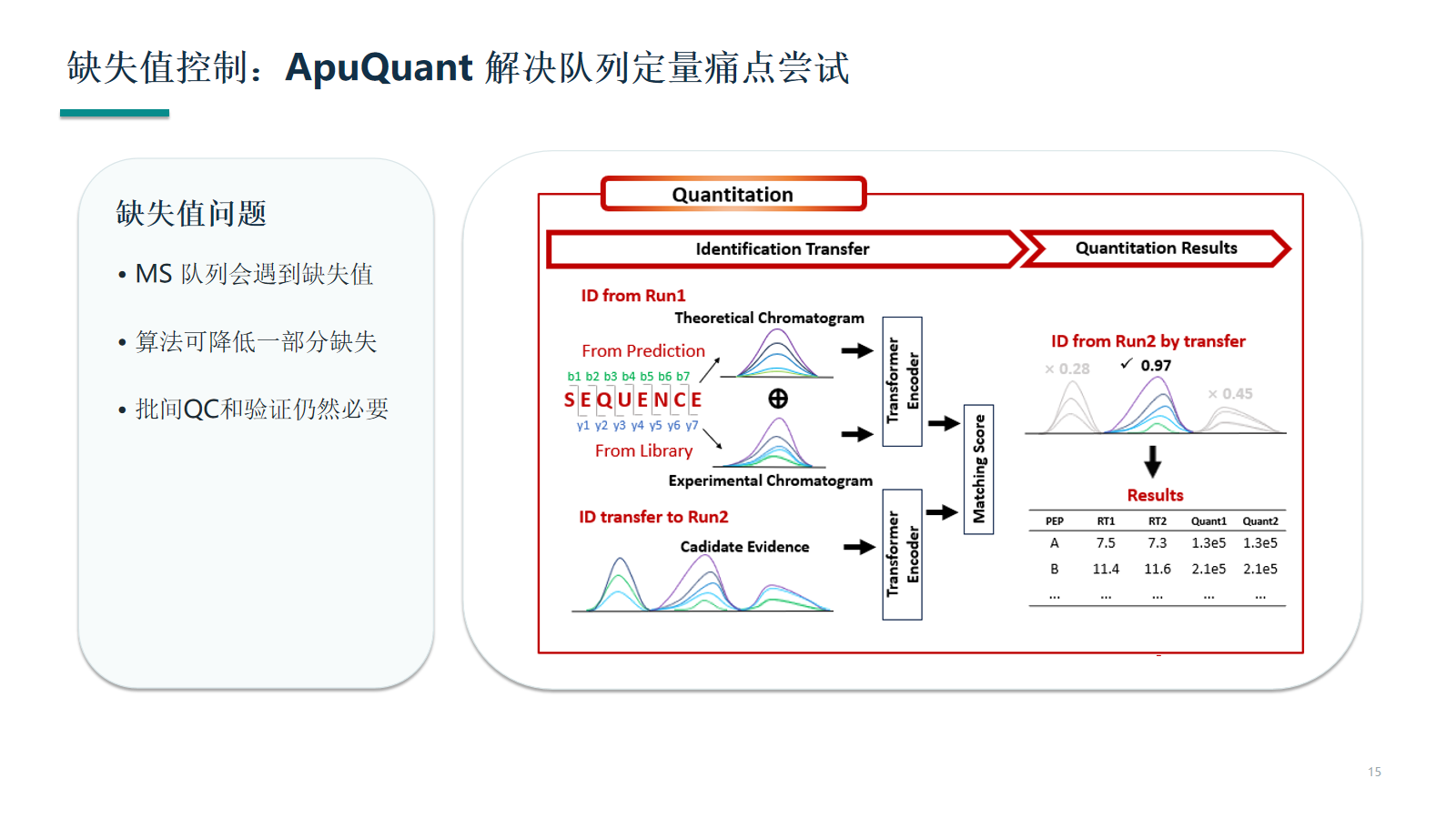
资料拆解
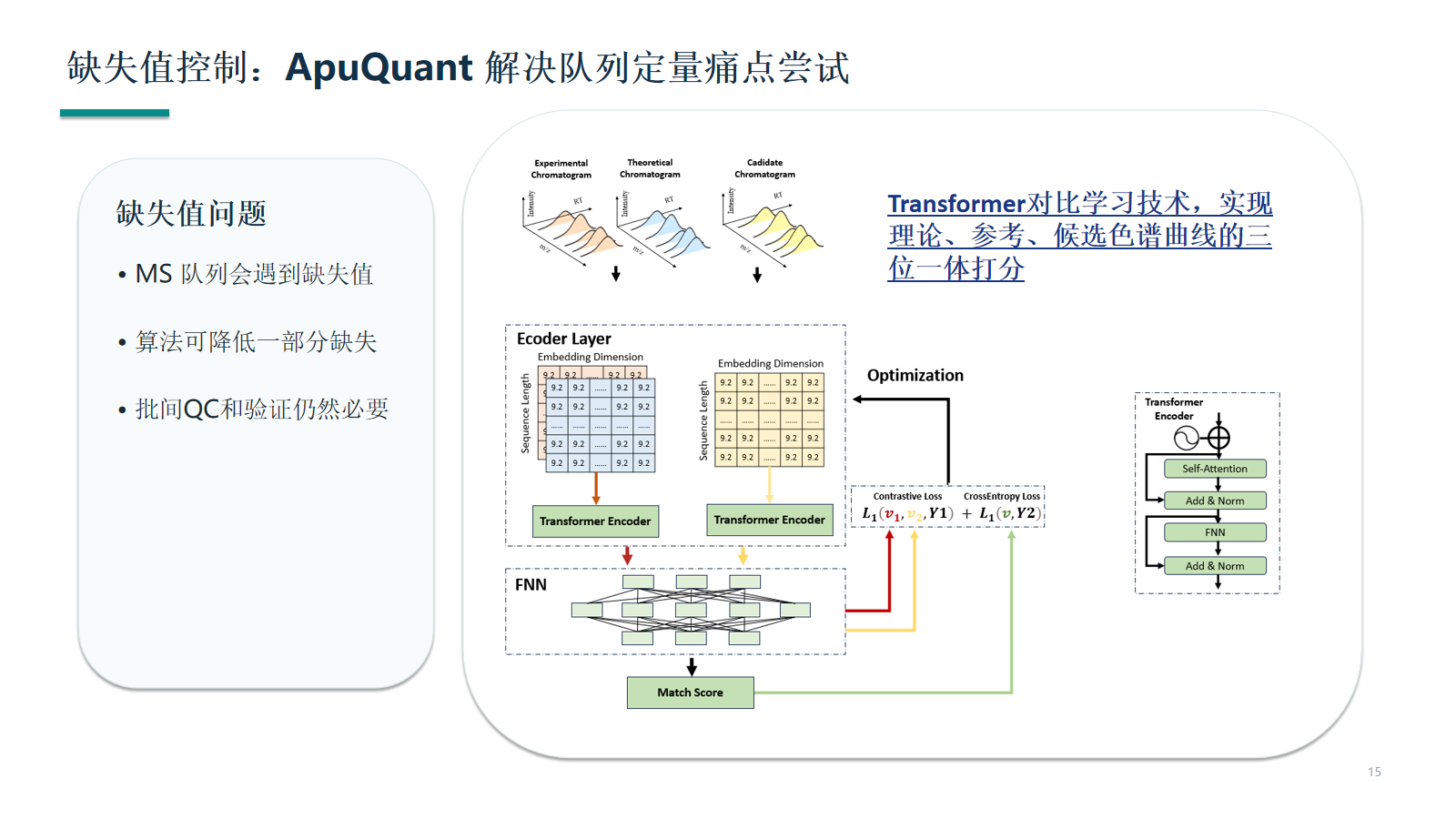
缺失值控制:ApuQuant 解决队列定量痛点尝试
Transformer 对比学习等方法可通过理论、参考和候选色谱曲线联合打分提升队列定量一致性。它的价值在于辅助峰识别和缺失控制,不应替代 QC 判断。
- 理论、参考和候选色谱曲线联合打分可提高峰识别一致性。
- Transformer 对比学习的输出仍应回到谱图、峰形和 QC 图中复核。
- 算法结论需要和标准肽、pooled QC、批次趋势共同解释。
查看原图
上机前风险
血浆样本有哪些上机前风险?
血浆动态范围跨越 mg/mL 到 pg/mL;采血管、凝血/抗凝、离心、冻融、保存和运输都会改变读数。前分析变量如果不控制,后续统计差异很容易失真。
- 同一项目内血浆和血清不应混用,病例/对照的采血和离心条件必须一致。
- 白蛋白、IgG、补体和凝血蛋白会通过质量占比、共流出和离子抑制影响低丰度信号。
- 血红蛋白、血小板和凝血相关 marker 应作为污染监测指标。
- 高丰度去除、PCA-N、磁珠富集和 EV 富集都会重构蛋白空间,必须验证偏倚是否可控。
动态范围阶梯
血浆蛋白浓度可跨越约 10 个数量级。条形用于提示数量级差异,不代表线性浓度比例,重点帮助判断低丰度候选物为什么容易被高丰度背景掩盖。
血浆信号空间主体,占据绝大多数质量负荷;未处理样本中会主导离子流和谱图解释。
仍属于高丰度背景,常与目标肽段共流出;多个肽段信号重叠后,软件更难准确拆分和定量。
跨越中高丰度区,炎症状态、溶血、采血管和冻融差异会显著改变可解释性。
进入低丰度候选区,需要前处理、色谱分离、DIA 窗口和数据端 QC 一起降低背景影响。
与高丰度蛋白相差约 10^9-10^10 量级,通常需要富集、靶向验证或专门平台支持。
资料拆解
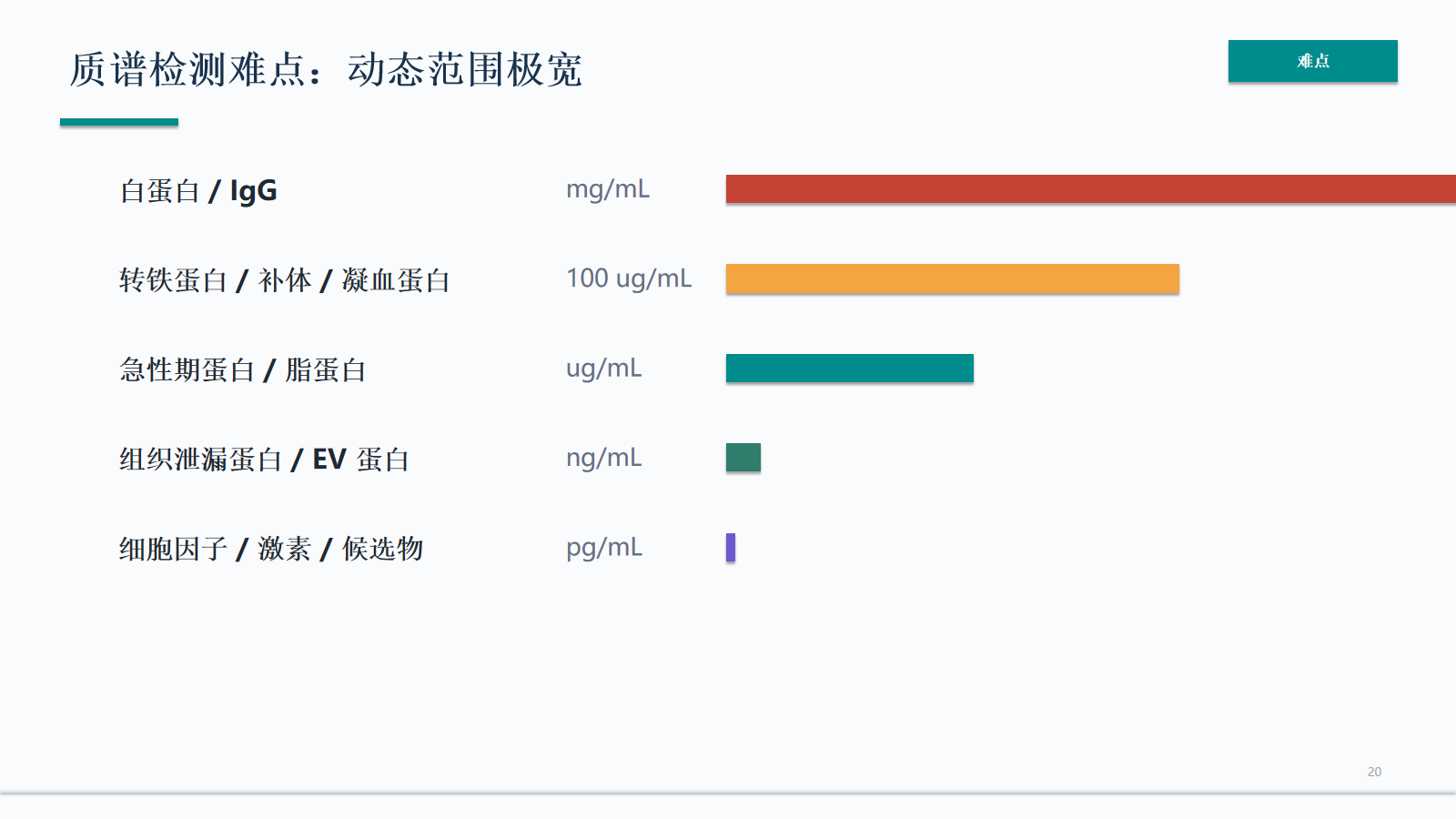
质谱检测难点:动态范围极宽
血液样本的动态范围跨越 mg/mL 到 pg/mL。白蛋白、IgG、补体和凝血蛋白会占据大量信号空间,使低丰度组织泄漏蛋白、EV 蛋白和细胞因子更难被 MS 稳定观察。
- 血液样本的动态范围跨越 mg/mL 到 pg/mL。白蛋白、IgG、补体和凝血蛋白会占据大量信号空间,使低丰度组织泄漏蛋白、EV 蛋白和细胞因子更难被 MS 稳定观察。
查看原图
资料拆解
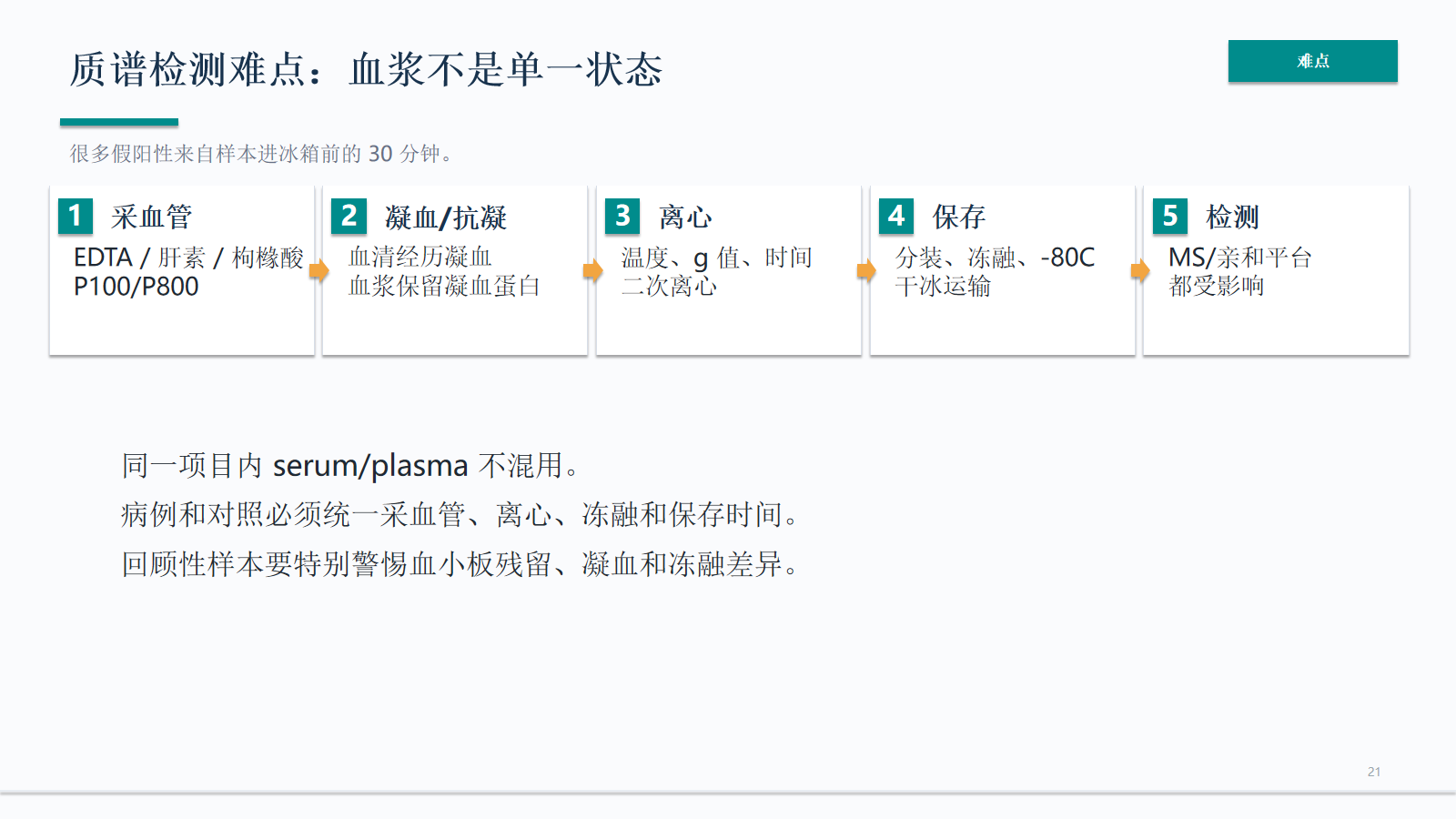
质谱检测难点:血浆不是单一状态
血浆不是单一状态。采血管、凝血/抗凝、离心、保存、冻融和运输都会改变样本读数;同一项目内 serum/plasma 不应混用,病例和对照也必须统一前分析条件。
- 血浆保留凝血蛋白
- 病例和对照必须统一采血管、离心、冻融和保存时间。
- 回顾性样本要特别警惕血小板残留、凝血和冻融差异。
查看原图
资料拆解
高丰度蛋白如何干扰低丰度蛋白
高丰度蛋白会通过质量占比、共流出、离子抑制、采集竞争、载体结合和污染放大干扰低丰度检测。统计差异可能来自采样偏差,而不是疾病生物学。
- 高丰度蛋白会通过质量占比、共流出、离子抑制、采集竞争、载体结合和污染放大干扰低丰度检测。统计差异可能来自采样偏差,而不是疾病生物学。
查看原图

资料拆解
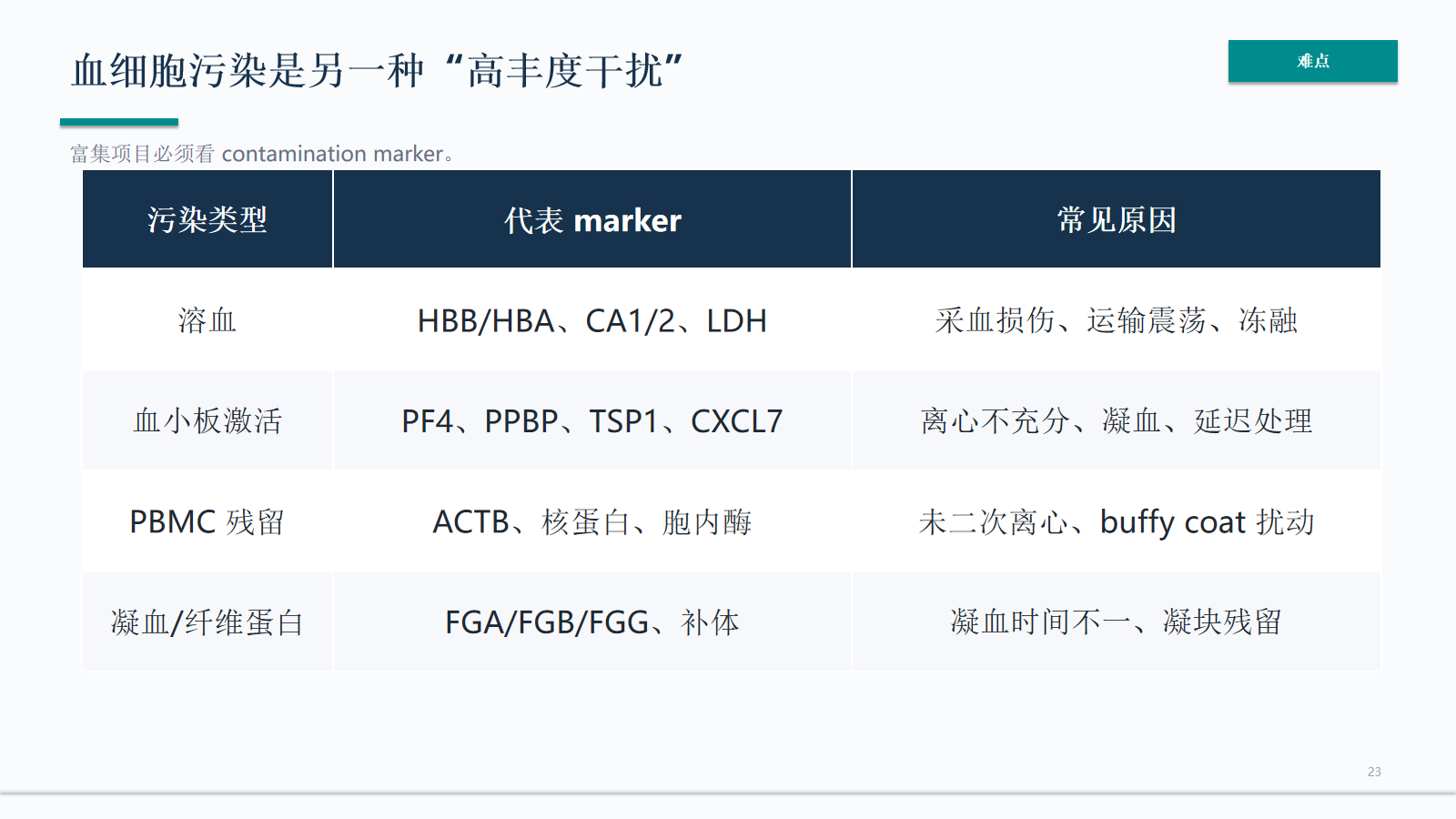
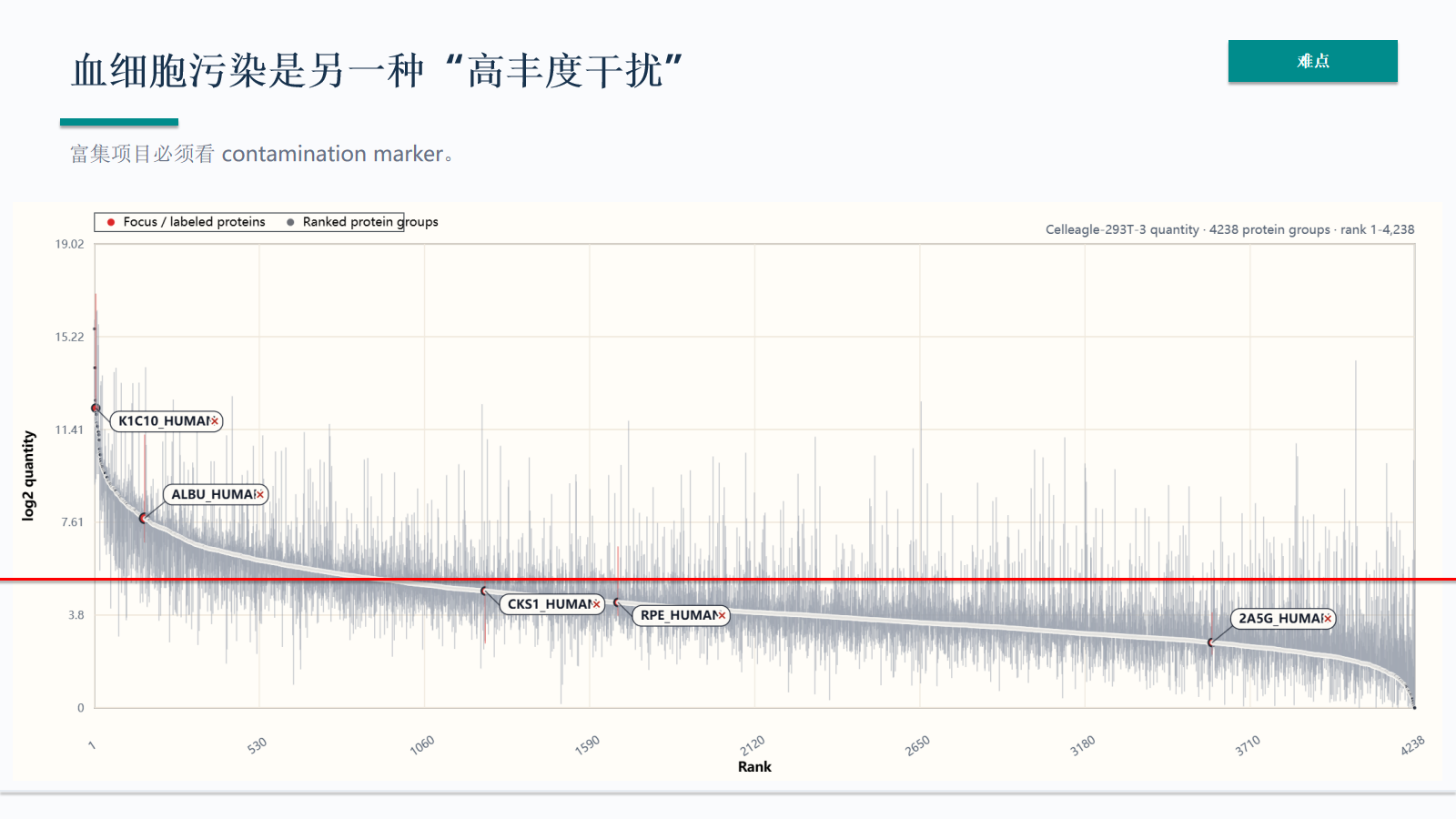
血细胞污染是另一种“高丰度干扰”
血细胞污染是血液 MS 中重要的高丰度干扰。血红蛋白、血小板和凝血相关 marker 可以帮助识别采血、离心、冻融或富集流程异常。
- 富集项目必须看 contamination marker。
查看原图
资料拆解
血细胞污染是另一种“高丰度干扰”
污染 marker 不只用于排除样本,也用于定位问题来源。若污染信号与分组、批次或采血地点相关,后续差异分析需要重新评估。
- 富集项目必须看 contamination marker。
查看原图
资料拆解
高丰度去除与选择性富集
高丰度去除、PCA-N、磁珠富集和 EV 富集都能改变可观测蛋白空间。关键不是单纯增加深度,而是判断偏倚是否可控、是否可复现、是否适合研究问题。
- 降低 albumin/IgG 等高丰度背景
- 低成本、低样本量、适合大队列 MS
- 提高低丰度覆盖、候选物多
查看原图

资料拆解
磁珠富集/纳米磁珠富集富集:适用边界
P2/Seer/易肽类路线属于 protein corona 选择性采样。它们不是无偏完整血浆,而是通过特定材料表面观察血浆蛋白空间的另一种窗口。
- P2/Seer/易肽 类路线是 corona 采样,不是无偏完整血浆。
查看原图

低丰度富集
低丰度富集如何接入自动化和验证?
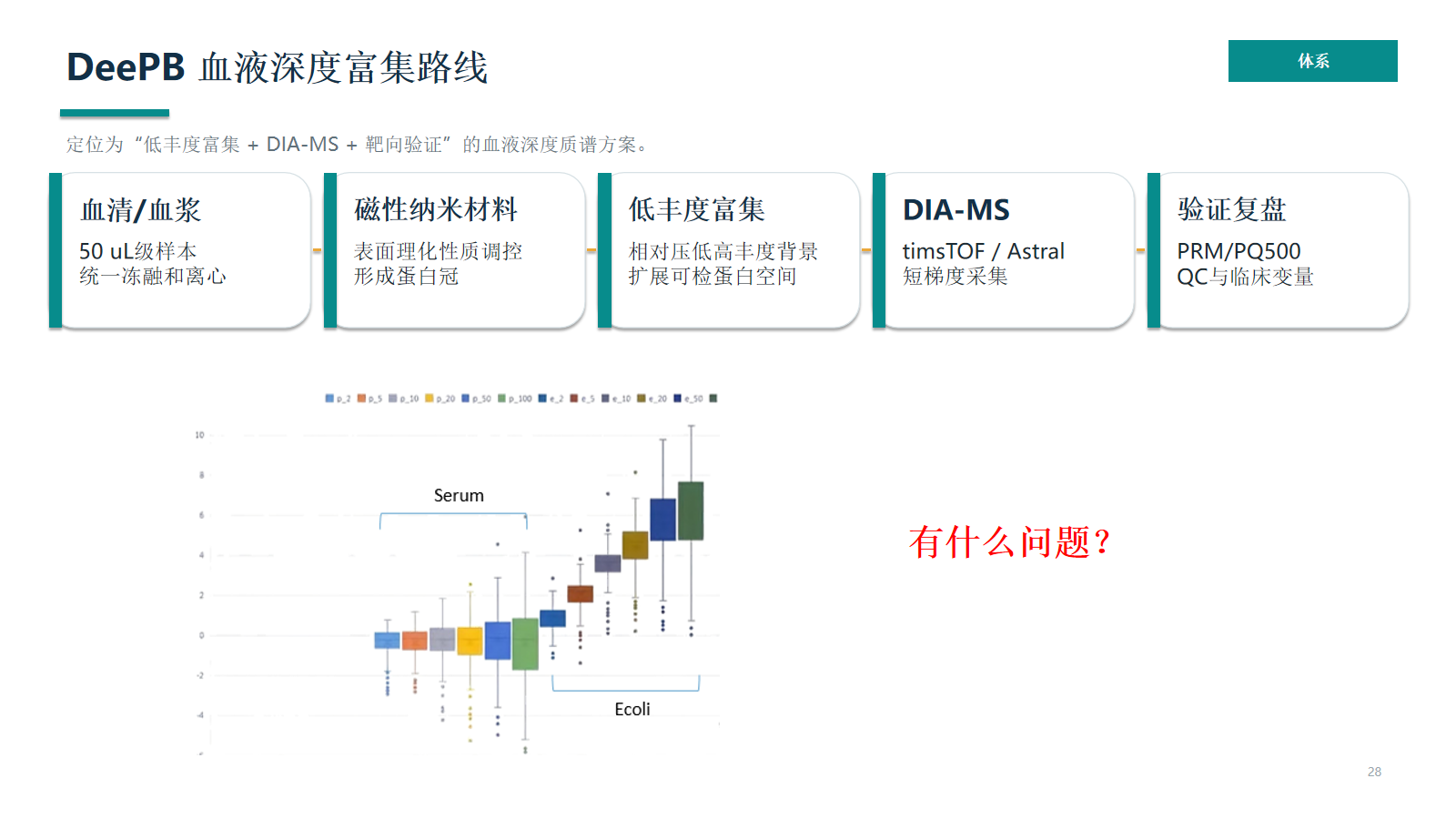
DeePB 血液深度富集路线把血清/血浆、磁性纳米材料、选择性采样、DIA-MS、PRM/PQ500 和 OmicsCloud QC 串成闭环。OSFP0002-96X 可作为易肽 DeeP 低丰度蛋白富集及前处理制备试剂盒的产品入口,用于把前处理步骤标准化。
- 样本输入、材料暴露、蛋白冠形成、洗脱酶解和 DIA-MS 采集要在同一 SOP 下运行。
- 自动化记录板位、试剂 lot、程序版本、反应时间和异常事件,降低系统性人为偏移。
- 性能数据应在具体样本类型、仪器、批次和 QC 标准下解释,不作为简单平台排名。
- PQ500 等重标肽资源可把 discovery 候选接入定性、校正和半绝对定量验证。
血清 / 血浆
50 µL 级样本,统一冻融、离心和采血管信息。
磁性纳米材料
通过表面理化性质形成蛋白冠,调节可进入 MS 的蛋白空间。
选择性采样
相对压低高丰度背景,改变可观测蛋白空间并扩展候选发现窗口。
DIA-MS
timsTOF / Astral 等平台可用于短梯度 discovery。
验证复盘
PRM/PQ500、QC 和临床变量共同决定候选物是否可解释。
样本端血清/血浆、采血管、冻融、离心、体积和异常记录
自动化端板位、试剂 lot、程序版本、反应时间和脱盐/富集记录
质谱端DIA-MS discovery、PRM/PQ500 验证、标准肽和长期 pooled QC
数据端污染 marker、缺失率、CV、PCA/UMAP、批次和项目复盘
资料拆解
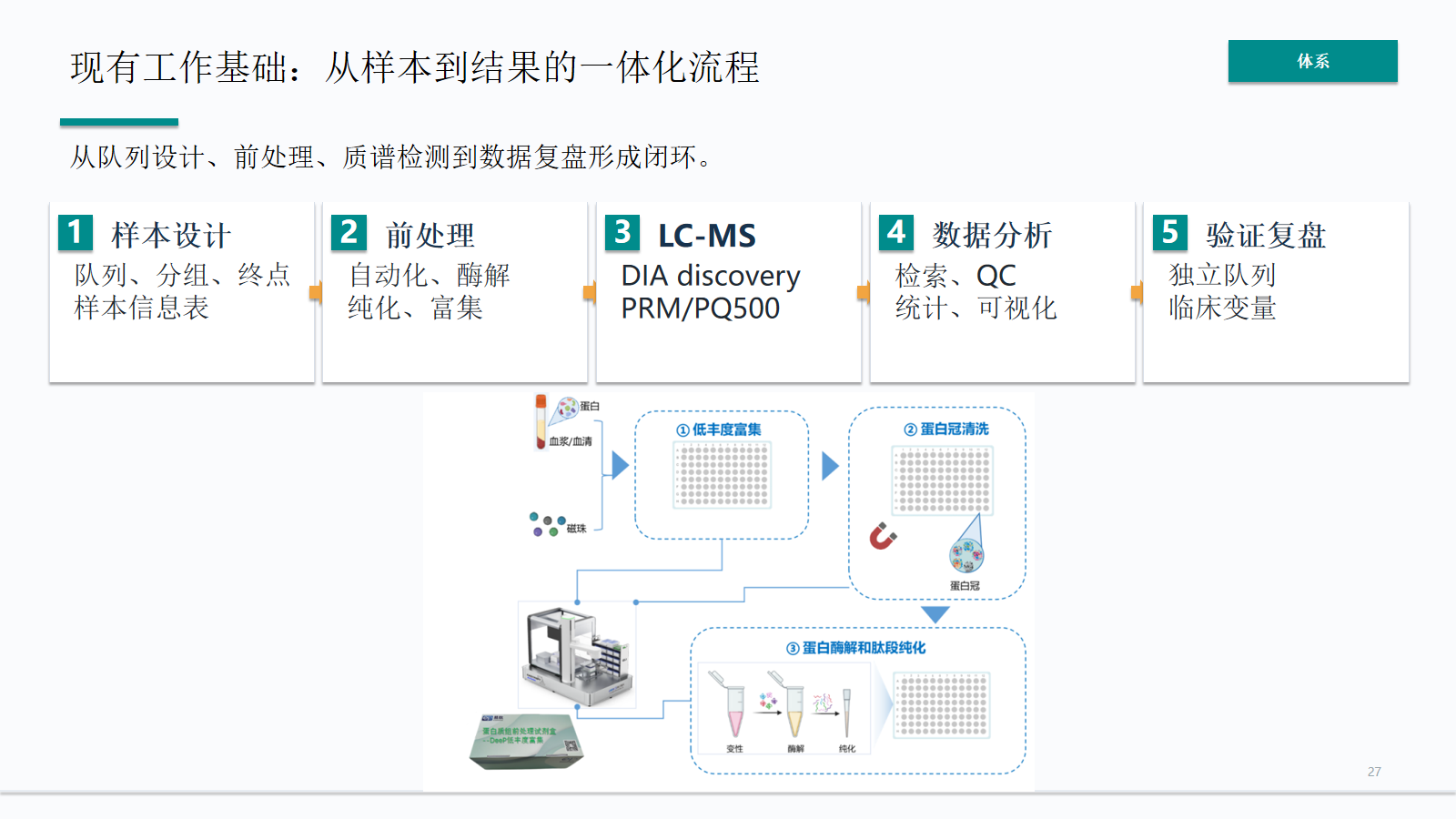
现有能力基础:从样本到结果的一体化流程
血液项目需要从队列设计到验证复盘形成闭环。样本信息表、前处理程序、LC-MS 采集、检索 QC、统计可视化和独立队列验证缺一不可。
- 从队列设计、前处理、质谱检测到数据复盘形成闭环。
查看原图
资料拆解
DeePB 血液深度富集路线
DeePB 路线把血清/血浆、磁性纳米材料、选择性采样、DIA-MS 和 PRM/PQ500 验证连接起来。它的目标是扩展可观测低丰度候选空间,同时保留 MS 证据链。
- 定位为“选择性富集 + DIA-MS + 靶向验证”的血液深度质谱方案。
查看原图
资料拆解
DeePB 性能数据:深度、速度和体液适用性
早期性能数据适合说明工作基础和可行性,不能直接作为外部平台横向排名。正式项目仍需按样本类型、批次设计、仪器平台和 QC 标准重新验证。
- 这些数据适合作为现有能力基础,不作为外部平台横向排名。
查看原图

资料拆解
DeePB 性能数据:稳定性、跨物种和PQ500衔接
深度富集方案必须同时证明稳定性、跨物种适配和验证路径。PQ500 等重标肽资源可把 discovery 候选进一步接入定性、校正和半绝对定量。
- 深度富集必须同时证明稳定性和验证路径。
- 人源血液关键蛋白;用于
查看原图

资料拆解
自动化为什么重要
自动化的核心价值是降低系统性人为偏移。裂解、还原烷基化、酶解、纯化、富集、脱盐、板位和试剂 lot 都应进入可审计记录。
- 板位、试剂 lot、程序版本、反应时间和异常记录
- pooled QC、标准肽、桥接样本和长期趋势图
- 适合研究队列
查看原图

资料拆解
样品制备:从传统长流程到快速标准化
快速标准化样本制备可减少人员差异和批次漂移。长流程并不一定更可靠,关键是每一步是否可控、可复核、可长期运行。
- 快速、标准化的样本制备降低人员差异和批次偏移。
查看原图

资料拆解
长期测试与QC
长期 QC 要同时看板间相关性、定量 CV 和异常样本识别。单次数据好看并不代表平台稳定,长期 pooled QC 和趋势图更能反映运行健康度。
- 适合观察长期漂移
查看原图

结果放行
结果进入分析前,QC 是否合格?
血液项目不能只看 protein groups。富集前样本是否合格、富集后污染和回收是否稳定、数据端批次是否可控,以及送样信息是否完整,都会决定结果能否解释。
- 富集前检查样本外观、离心质量、纤维蛋白、细胞碎片、冻融次数和输入体积。
- 富集后检查污染 marker、标准肽、回收线性、缺失率和跨批次稳定性。
- OmicsCloud 可集中查看 CV、PCA/UMAP、缺失率、污染 heatmap 和项目批次。
- 送样记录应包含采血管、离心、分装、冻融、运输、异常记录和样本信息表。
富集前
离心、血细胞残留、纤维蛋白、冻融、样本体积和异常外观。
富集中
去除柱/磁珠容量、输入量、洗脱备份、流穿备份和批次记录。
富集后
标准肽、污染 marker、回收线性、可观测空间扩展和跨批次稳定性。
数据端
PCA/UMAP、pooled QC CV、缺失率分布、Hb/PF4/PPBP/FGA/FGB/FGG heatmap。
资料拆解
高丰度去除:操作风险点
高丰度去除和富集操作要关注容量限制、样本离心、个体差异、保留备份和 co-depletion。异常样本需要能回查原样、流穿和洗脱。
- 样本必须严谨离心,避免纤维蛋白和细胞碎片影响 beads富集
- 去除柱/磁珠有容量限制,输入量不能超载。
- 病例/对照不应差异过大,个体间差异大也会影响富集特异性。
- 保留原样、流穿和洗脱备份,用于异常回查。
- 被 albumin/IgG 携带的蛋白可能发生 co-depletion。
查看原图
资料拆解
富集前 QC 标准
富集前 QC 决定后续数据是否值得解释。样本外观、离心质量、纤维蛋白、细胞碎片、冻融和输入体积都应在上机前记录。
- 富集前先记录样本外观、离心质量、纤维蛋白、细胞碎片和冻融次数。
- 确认输入体积、采血管、保存时间和异常记录后再进入富集流程。
- 不合格样本应优先暂停或标记,不应直接进入差异解释。
查看原图

资料拆解
富集后 QC 标准
富集后 QC 不能只看 protein groups。污染 marker、回收线性、标准肽、可观测空间扩展、批间稳定和缺失率共同决定结果可信度。
- 富集后同时检查污染 marker、protein groups、可观测空间扩展和缺失率。
- 标准肽、pooled QC 和桥接样本用于评估回收和跨批次稳定。
- 异常结果需要回查原样、流穿、洗脱和板位记录。
查看原图
资料拆解
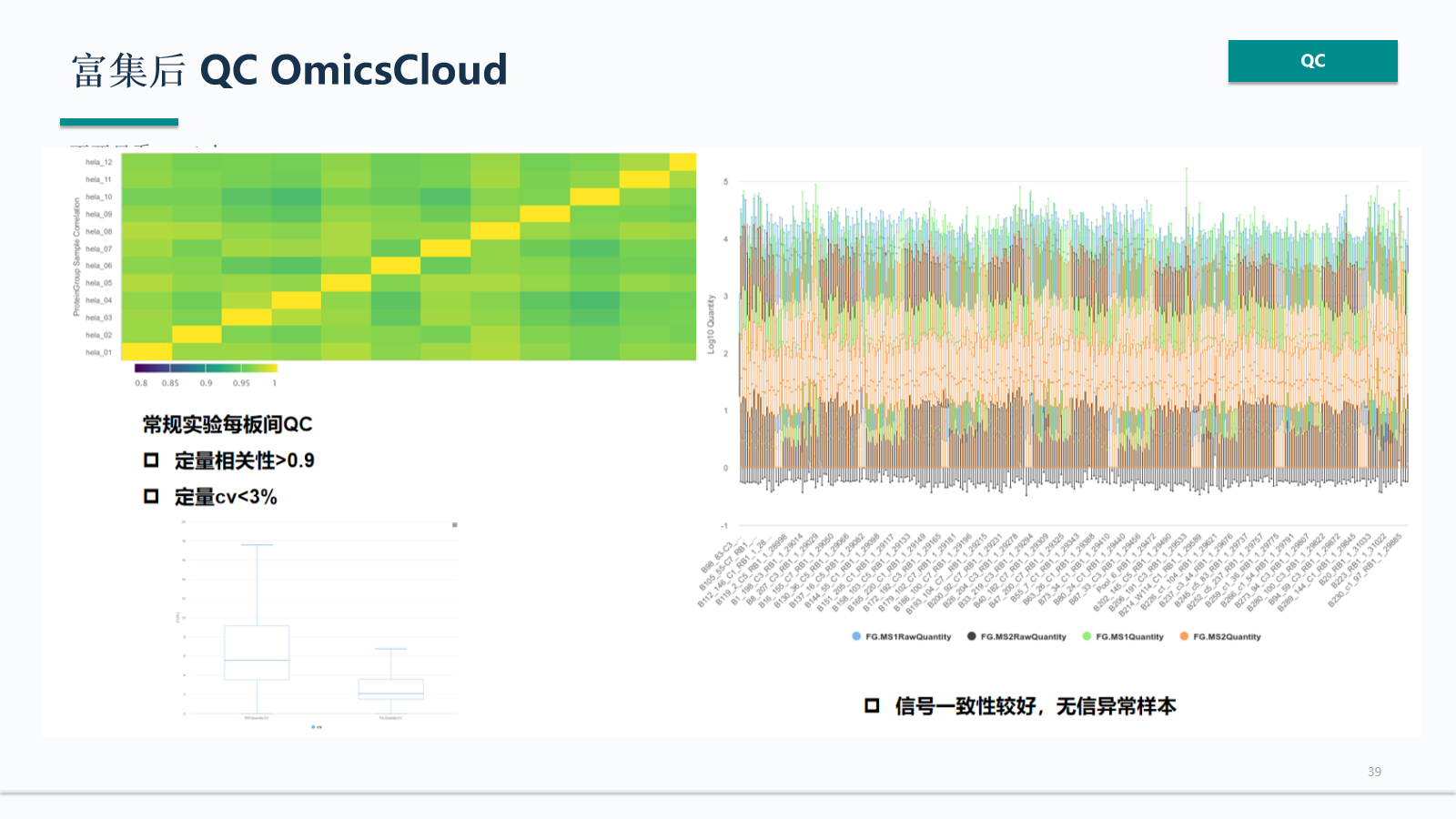
富集后 QC OmicsCloud
OmicsCloud 可承接富集后 QC 的集中查看:把污染、缺失、CV、PCA/UMAP 和项目批次放在同一处,帮助判断结果是否可进入统计分析。
- OmicsCloud 可集中查看污染、缺失、CV、PCA/UMAP 和项目批次。
- QC 结论应决定结果是否进入差异分析,而不是只作为附图展示。
- 富集前后的 QC 记录应与样本表、批次表和质谱运行记录关联。
查看原图
资料拆解
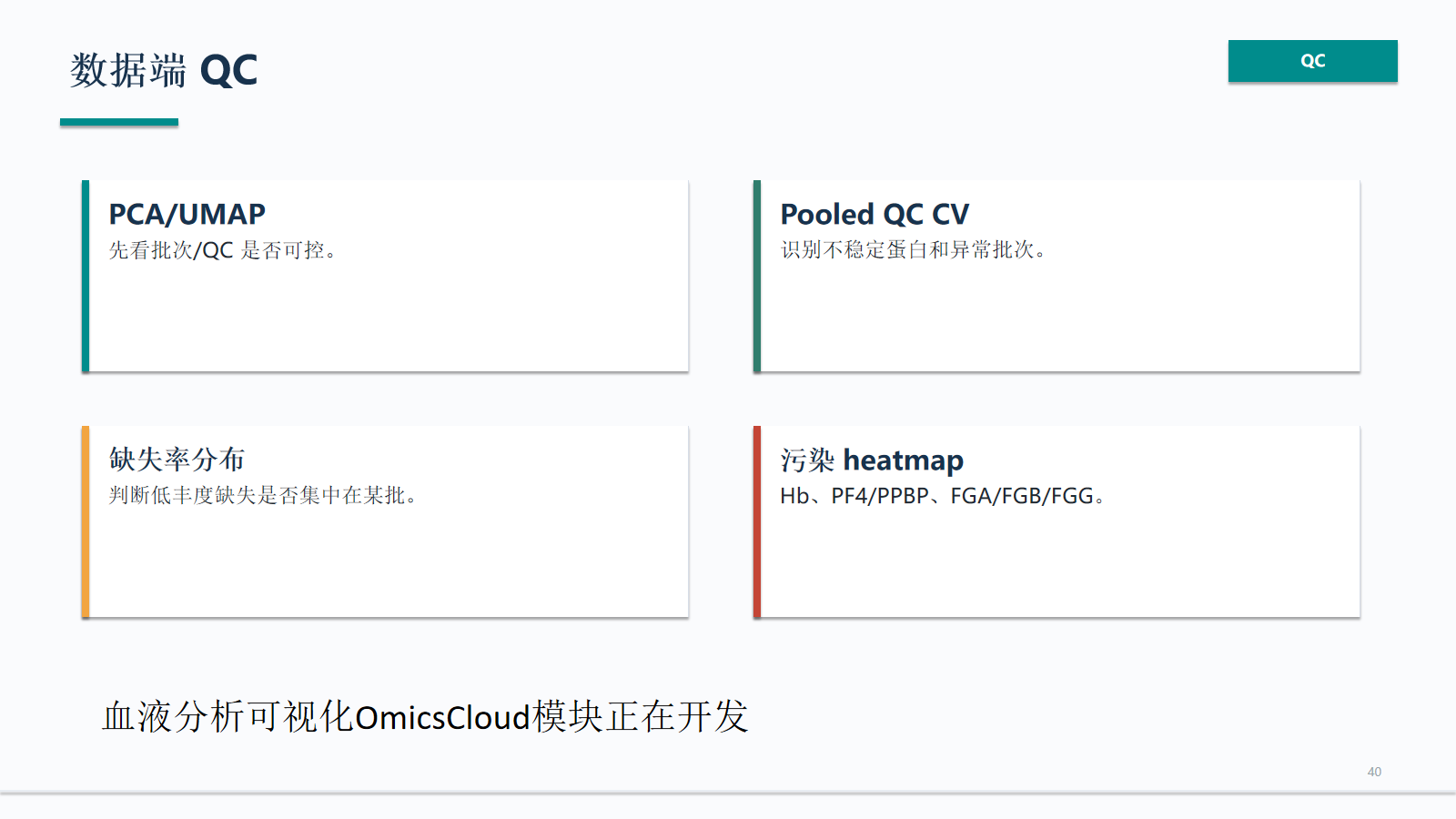
数据端 QC
数据端 QC 应先排查批次/QC 是否可控,再进入差异和功能解释。PCA/UMAP、pooled QC CV、缺失率分布和污染 heatmap 是血液项目的基础图谱。
- 判断低丰度缺失是否集中在某批。
- Hb、PF4/PPBP、FGA/FGB/FGG。
- 血液分析可视化可在 OmicsCloud 模块中承接。
查看原图
资料拆解
样本保存和送样规范
送样规范决定数据是否可解释。采血管、离心、分装、冻融、运输、样本信息和异常记录都属于数据链条的一部分。
- 送样不是把管子寄出去,而是把可解释的数据链条一起交出去。
查看原图

交付判断
最终交付的是可解释证据,不是蛋白数量
血液蛋白质组学的稳健路线不是单一平台,而是样本 SOP、自动化前处理、LC-MS 或亲和平台、靶向验证、数据 QC 和交付复盘的整体组合。
- 发现阶段要广,验证阶段要准,研究评估阶段要能解释、能复现。
- 深度必须受 QC 约束,不能用蛋白数量替代污染、缺失和批次稳定性判断。
- 面向医院检测平台时,应优先建设可审计流程、可复核数据和可持续交付能力。
资料拆解
现有能力基础
现有能力基础应理解为平台化能力组合:血液前处理、自动化、DIA-MS、PRM/PQ500、OmicsCloud QC 和项目复盘共同支撑血液项目。
- 现有能力基础应理解为平台化能力组合:血液前处理、自动化、DIA-MS、PRM/PQ500、OmicsCloud QC 和项目复盘共同支撑血液项目。
查看原图

资料拆解
总结
血液蛋白质组学的核心是把复杂样本变成可解释证据。深度必须受 QC 约束,发现、验证和研究评估需要整体设计。
- 血液蛋白质组学的核心是把复杂样本变成可解释证据。
- 血液适合早筛、分型、
- 2. 深度必须受 QC 约束
- 去除和富集能提高覆盖,
- 但必须以污染指数、标准肽
查看原图

资料来源
可继续外链阅读的资料
以下资料用于扩展平台背景、产品参数和公开队列研究语境。正文判断以本页学习路径为主,外链用于补充核验和继续阅读。
易肽 OSFP0002-96X易肽 DeeP 低丰度蛋白富集及前处理制备试剂盒,规格 96X,可作为血液 DeePB/选择性富集前处理的产品入口。
Olink Explore HTPEA、约 5,400 蛋白、2 µL plasma/serum、2000+ samples/week 等官方参数。
SomaScan 11KSomaScan 11K 平台与多样本类型扩展的官方新闻资料。
NULISA Inflammation AQ250+ biomarker、150+ 绝对定量目标、fg/mL 灵敏度和动态范围信息。
Seer Proteograph蛋白冠/纳米颗粒路线、up to 10x protein groups 和 1000+ samples/week 语境。
UK Biobank plasma proteomics54,219 例 UK Biobank plasma proteomics、pQTL 和健康关联研究。
Human Blood Atlas血液蛋白、免疫细胞、分泌蛋白和血液图谱背景。