交付层的核心作用
平台化交付需要同时建立 schema、字段词典、QC 历史、pipeline 结果留存与 XIC 回查能力。
报表与 QC
Spectronaut 报表部分覆盖 Report Perspective、QC Perspective、Pipeline、
关键报表字段 与 XIC 导出数据库,对应
Spectronaut 的交付、复核与长期运行主线。
总述
如果分析流程负责把数据分析出来,那么报表、QC 和 Pipeline 负责把结果变成能复核、能交付、能长期运行的体系。附录 8 和 9 则进一步把字段语义和 chromatogram 证据向外打开。
平台化交付需要同时建立 schema、字段词典、QC 历史、pipeline 结果留存与 XIC 回查能力。
从分析结果到 schema、字段对象、QC、pipeline 和 XIC 接口的工作链。
报表结构
报表结构由 schema tree、column chooser、filters 与 report preview 四个核心面板构成,分别对应结构、字段、筛选与输出预演。
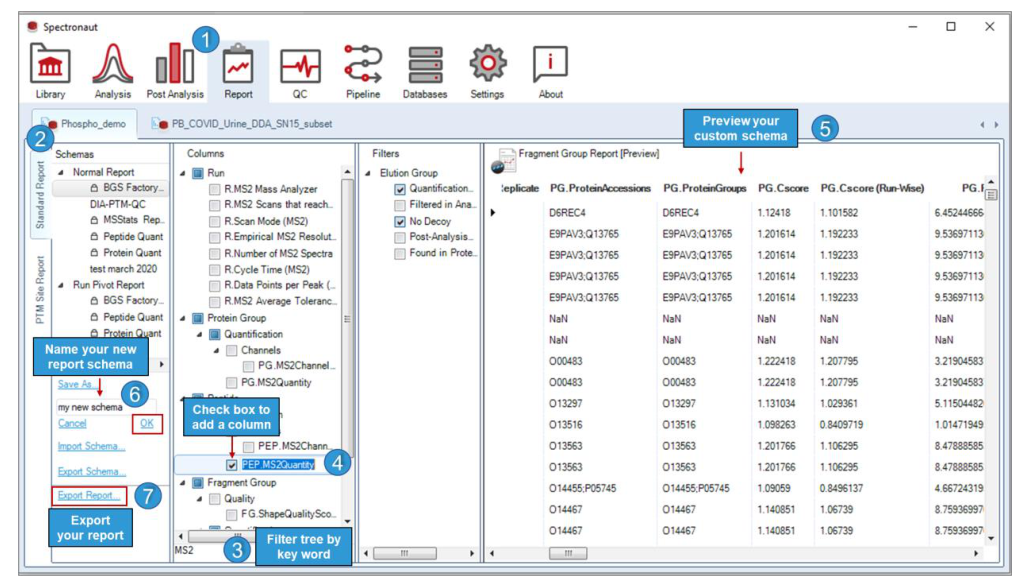
Schema tree 定义报表结构,Column chooser 负责选字段,Filters 控制筛选条件,Report preview 对应导出前的结构预演。读取顺序为先对象层、再字段层、再筛选层。
Normal Report 主要对应统计和数据库入库,Run Pivot 对应人工浏览,PTM Site Report 对位点项目尤其重要,Grid View Report 对应界面视图联动。
QC
QC 不只是展示当前结果,而是持续记录成功分析、按 instrument 构建历史,并允许样本特异 QC panel 与 iRT QC 并行存在。
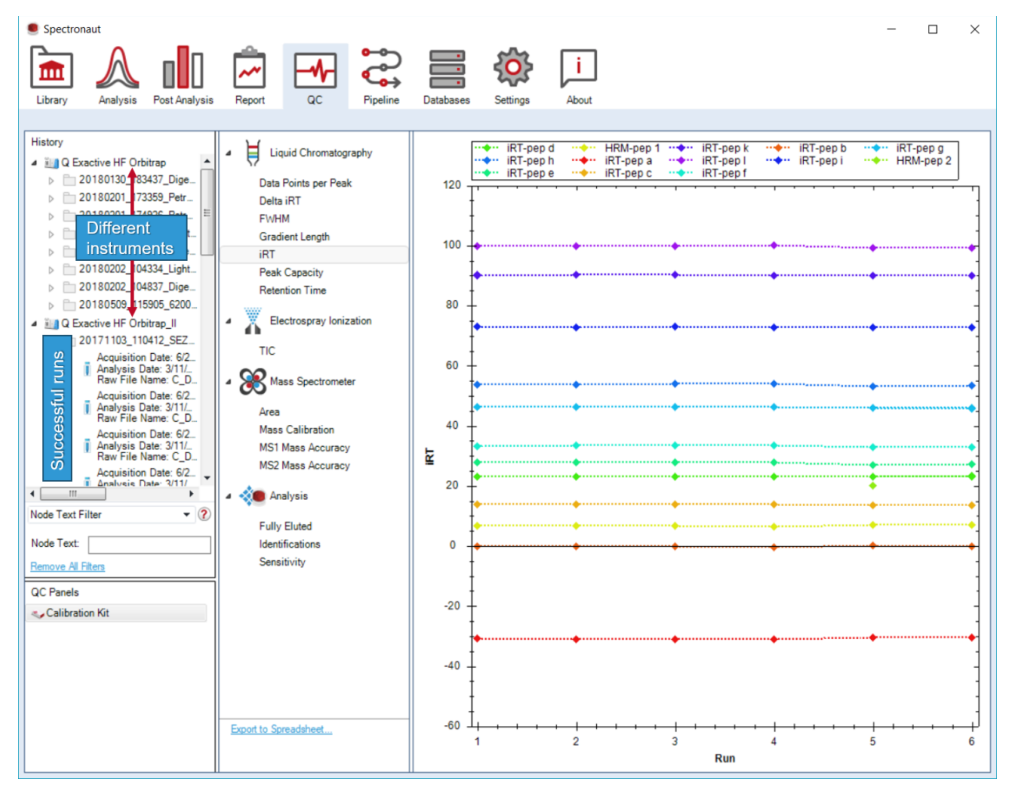
Enable QC 生成 sample-specific QC panel。QC Plot History Length 决定历史窗口大小,通常按实验室管理策略统一设置。QC Plot History Length 控制 plots 中显示多少历史 runs,这意味着 QC 不是“自动无限堆叠”,而是一个需要人为定义观察窗口的监控系统。Enable QC 后,还能把特定 spectral library 派生出 sample-specific QC panel。这一步说明 discovery 文件可以反向服务平台质控。Pipeline
Spectronaut 更偏向顺序处理多个实验,而不是并行抢占磁盘 IO。Pipeline 的核心是稳定、可复制和批量运行。
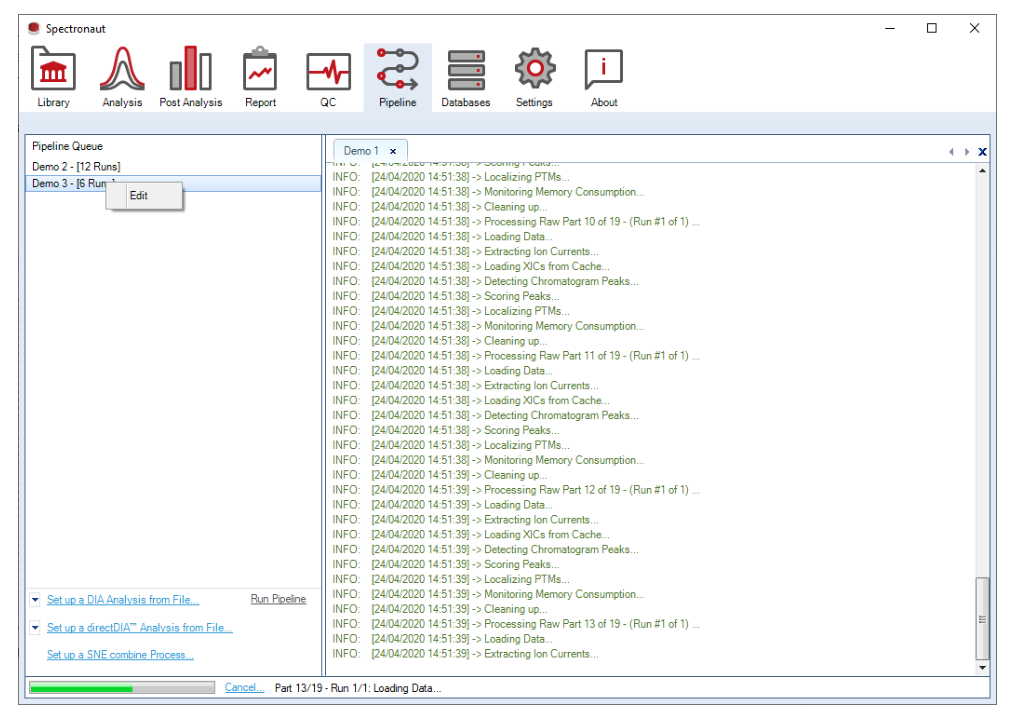
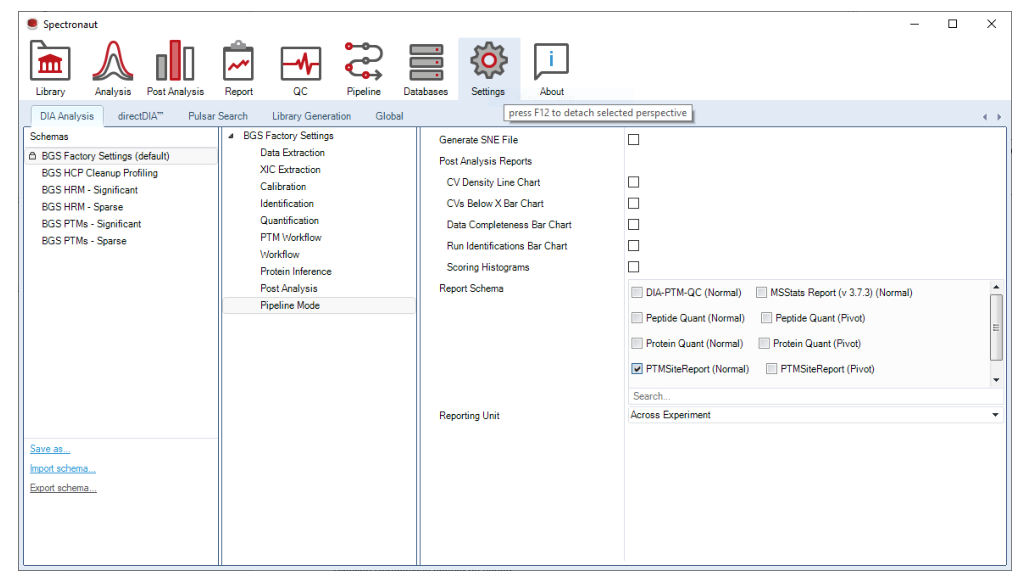
.sne。Pipeline 不是为了替代所有人工 review,而是为了让标准化 discovery 项目进入队列式批处理,减少重复点击与输出不一致。
这一层决定写出哪些 reports、按 run 还是按 experiment 组织、是否导出 scoring histograms,以及是否保留整个实验的 .sne 文件,是平台交付设计的关键开关。
.sne,再在 combine 阶段以 FDR-controlled 方式合并 identification results,并重新做 cross-run normalization、interference correction、protein inference、quantification 和 FDR。附录 8
这一部分把 report 输出从列名列表提升到对象层级语言,可直接作为字段词典起点。
EG.Qvalue 对应 precursor / elution group 层,需要和峰形、iRT 偏移、fragment 干扰一起判断;PG.Qvalue 对应 protein group 层,更接近最终交付对象。两者都叫 q-value,但回答的并不是同一个问题。
Protein group 不是 precursor 结果的自然终点,而是经过 protein inference、shared peptides 处理和单 hit 蛋白策略之后的交付对象。只要这些规则变化,PG.Qvalue、蛋白数和最终可报告边界就会一起变化。
因此蛋白层结果不能只说“我们做了 1% FDR”。更完整的表达应同时说明:蛋白层是如何从 precursor / peptide 汇总而来,single-hit proteins 是否被保留,以及当前设置在 entrapment 或经验验证中落到了什么风险水平。
| 层级 | 代表字段 | 解释重点 | 主要使用场景 |
|---|---|---|---|
| PG | PG.Quantity、PG.Qvalue、PG.RunEvidenceCount |
回答蛋白层对象是否可靠、定量值是多少、每个 run 的支持证据有多强。 | 结果汇报、差异蛋白解释、项目交付 |
| PEP | PEP.GroupingKey、PEP.IsProteinGroupSpecific、PEP.UsedForProteinGroupQuantity |
这个肽段如何被定义、是否特异、是否真的参与了蛋白层定量。 | 定量逻辑理解、蛋白推断理解 |
| EG | EG.PrecursorId、EG.Qvalue、EG.Cscore、EG.DeltaiRT、EG.MeanTailingFactor |
回答这个前体在这个 run 中的鉴定与峰提取到底可信不可信。 | 峰图 review、质控、排错 |
| FG / F | FG.TotalPeakArea、FG.Quantity、F.InterferenceScore、F.ExcludeFromQuantification |
说明定量是由哪些 fragment 累积而来,以及哪些离子可能被排除或视为干扰。 | 碎片离子层排错、labeled / spike-in 项目 |
| PTM | PTM.Quantity、PTM.QuantityPerProtein、PTM.Stoichiometry |
把位点项目从有无变化推进到相对蛋白输入的变化和占比解释。 | 磷酸化、乙酰化等 PTM 项目 |
| 交付场景 | 报告中至少同时读取的字段 | 报告里必须补充的方法说明 | 当前可以成立的结论 |
|---|---|---|---|
| 标准 canonical FASTA、默认 protein inference | PG.Qvalue、PG.RunEvidenceCount、PEP.UsedForProteinGroupQuantity |
数据库版本、protein inference 规则、single-hit protein 沿用默认设置 | 可以进入常规蛋白层结果汇报与差异分析 |
| canonical + non-canonical FASTA | PG.Qvalue、PEP.IsProteinGroupSpecific、PEP.GroupingKey、组别化 precursor 证据 |
数据库分组结构、precursor FDR per group、non-canonical 条目来源 |
蛋白层结果只能在当前数据库结构内解释,不能直接外推为标准蛋白组结论 |
| 保留 single-hit proteins | PG.Qvalue、PG.RunEvidenceCount、蛋白特异肽支持字段 |
single-hit protein 保留策略、经验验证方式、protein inference 版本 | 可以保留探索性蛋白层对象,但必须同步标注风险边界 |
| 开放搜索 / peptidomics / PTM probing | EG.Qvalue、PG.Qvalue、位点或肽段层证据字段 |
search space 范围、FDR 粒度、对象层级和过滤条件 | 通常先交付 discovery evidence,再谨慎收敛到蛋白层解释 |
PG.Qvalue 是 experiment-wise 的,所以不要把它直接理解成某个单一峰的质量。PEP.UsedForProteinGroupQuantity 直接说明这个肽会或不会进入蛋白定量。EG.Cscore、EG.DeltaiRT 和 EG.MeanTailingFactor 应与 XIC 和人工 review 一起讲。F.InterferenceScore 和 F.ExcludeFromQuantification 说明 fragment 级错误会向上游层级传导。PG.Qvalue 的解读边界也会随之改变,因此蛋白层字段必须和 settings 一起回看。protein inference。蛋白层对象不是 precursor 的自然相加,而是 shared peptides、grouping 和推断规则处理后的结果。single-hit protein 策略。是否保留仅由单条证据支持的蛋白,会直接改变蛋白数、经验错误比例和交付边界。PG.Qvalue 包装成统一的蛋白层结论。PG.ProteinGroups、PG.ProteinAccessions、PG.Genes、PG.ProteinDescriptions 都依赖 parsing rule,因此蛋白层字段好不好读,前提在于 Databases 里的 FASTA parsing 是否统一好。PEP.GroupingKey 与 PEP.GroupingKeyType 会告诉你当前 settings 把什么对象当成 peptide,默认通常是 stripped sequence。这直接决定 peptide 层的统计对象定义。EG.IsUserPeak、EG.IsVerified、EG.Cscore、EG.DeltaiRT、EG.MeanTailingFactor 需要与 review 行为对应起来阅读,因为它们会同时暴露峰是否被手改、峰是否可信以及色谱是否稳定。FG.Id、FG.TotalPeakArea、FG.Quantity 只在 labeled / spike-in workflows 下尤为关键,说明 fragment group 层并不是所有实验都同等重要。PTM.CollapseKey、PTM.Multiplicity、PTM.QuantityPerProtein 和 PTM.Stoichiometry,因为它们直接把 site collapse、input normalization 和 site stoichiometry 的逻辑写进报表字段里。Appendix 9
它让 Spectronaut 不再只是桌面软件,而成为可以和数据库、脚本和算法系统对接的数据接口层。
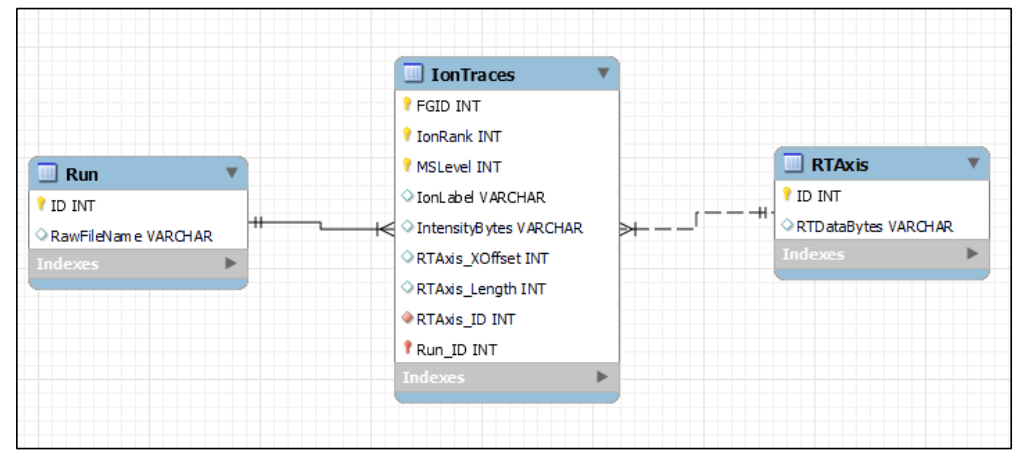
IonTraces、RTAxis 和 Run 三张表的关系。| 对象 | 作用 | 关键点 |
|---|---|---|
FG.XICDBID |
把 report 中的 feature / precursor 行回连到 XIC 数据库对象 | 它是报表层与色谱证据层之间最关键的桥 |
IonTraces |
保存强度轨迹和离子相关索引 | 峰形证据集中保存在这一层,而不是只停留在 report 的数量列中 |
RTAxis |
保存 RT 轴,减少重复存储 | 说明 XIC 可用数据库方式组织与索引 |
Run |
回溯 raw file 元信息 | 对应项目归档、异常追溯和跨 run 调试 |
Settings → Global → Reporting 启用 Automatic XIC Storage;或在 command line 的 Analysis Settings → Pipeline Mode 中启用 Export All XICs。IonTraces 是主表,FG.XICDBID 对应其 FGID,并携带 IonRank、MSLevel、IntensityBytes、RTAxis offsets 与 Run_ID;RTAxis 存整段 RT axis,再由 offset / length 截取子区间;Run 负责保存 source raw file 的文件名与索引。Skyline Schema
该 .skyr 是 Skyline report schema,用于把 Skyline 中的 precursor、fragment、iRT、修饰序列、蛋白和 decoy 信息导出成 Spectronaut library 可读字段。
schema 的 rowsource 指向 Transition,并展开 Results 子列表,因此它不是只导 peptide 列表,而是按 transition / fragment 级别输出 library 所需信息。
<report name="Spectronaut_Library" rowsource="...Transition" sublist="Results!*">
ProteinId、ModifiedSequence、StrippedSequence、iRT、PrecursorMz、PrecursorCharge、FragmentMz、FragmentCharge、FragmentType、FragmentNumber、RelativeFragmentIntensity、Decoy、IsotopicLabel 和 GeneId 是导出主干。
schema 同时过滤 Protein.Name != Decoys 与 Precursor.IsDecoy != TRUE,避免 decoy 对象进入目标 library 导出。
相关专题
Analysis、附录图谱和问题集分别承接 workflow、图谱复核与字段排错。