默认设置与版本一致性
因为 Spectronaut 的大部分项目稳定性问题,不是出在算法本身,而是出在路径规划、schema 版本、数据库版本、命名规则和报表约定上。只要这些没有统一好,每个分析师都会跑出自己的一套结果组织方式。
Spectronaut Settings
Spectronaut Settings 覆盖
Databases Perspective、Settings Perspective、File Name Parsing、
Command Line、Exit Codes、HTRMS Converter 和 BGMS Raw API。
这些部分共同定义数据库版本、默认模板、命令行参数与批处理接口。
Orientation
Databases、schema、directories、reporting、command-line 和 converter 决定的是整个实验室如何重复运行同一种 discovery 工作流,以及如何把它接入更大的批处理环境。
因为 Spectronaut 的大部分项目稳定性问题,不是出在算法本身,而是出在路径规划、schema 版本、数据库版本、命名规则和报表约定上。只要这些没有统一好,每个分析师都会跑出自己的一套结果组织方式。
从数据库、schema、命令行到 converter 的整体技术路径。
Databases
蛋白数据库、修饰库、酶切规则、GO 和表格导入被统一放在 Databases Perspective 里,这意味着这些并不是建库前临时准备的附件,而是软件长期维护的数据库和注释规则。
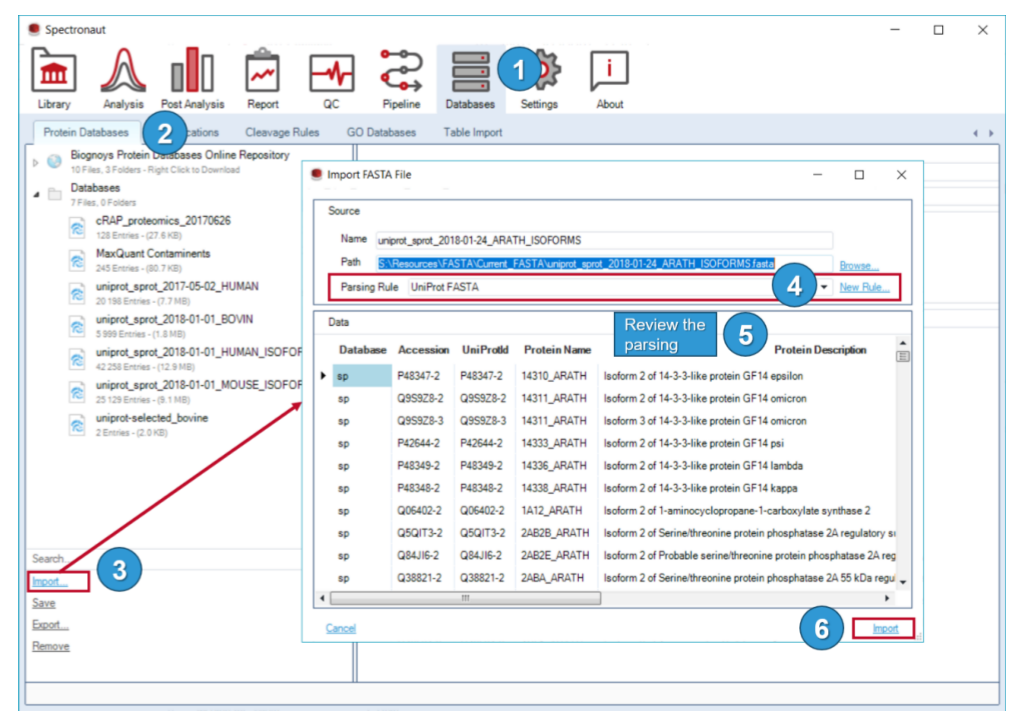
本模块固定三件事:FASTA 版本管理;解析规则对 accession、gene、description 等字段的定义;不同项目 FASTA 版本不统一时对后续 library 和 report 字段一致性的影响。
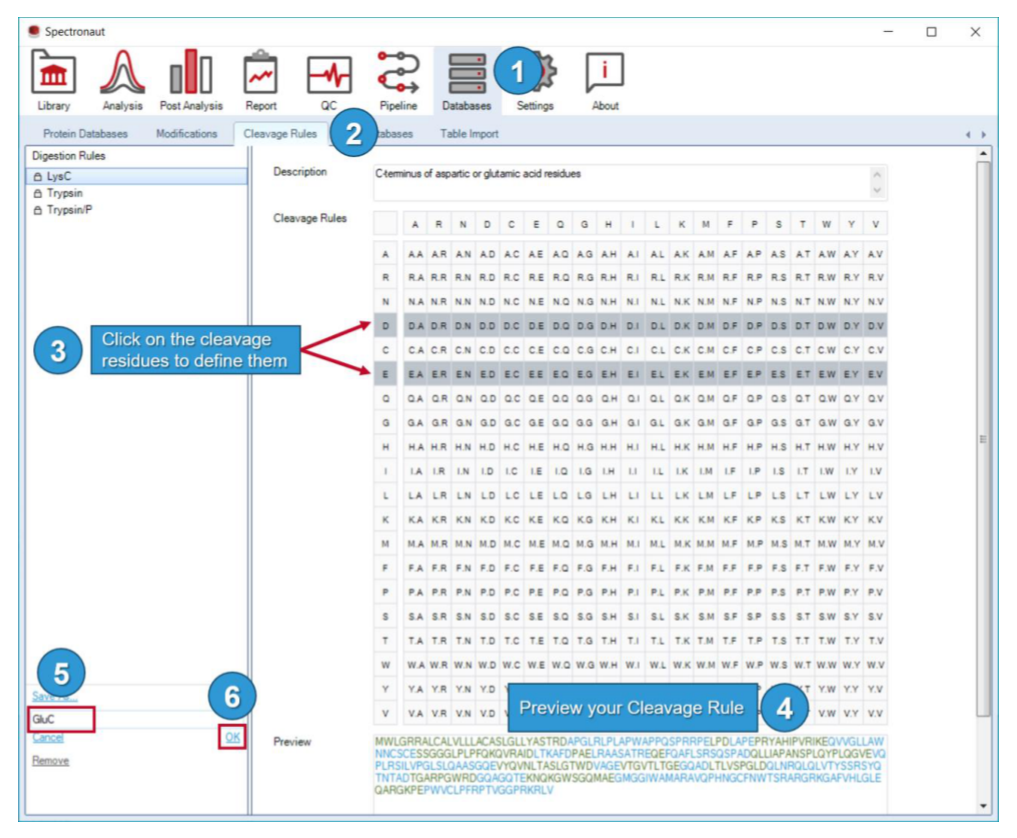
自定义 modification 主要服务 PTM 和化学生物学场景;cleavage rules 决定搜索空间大小和假阳性压力;GO database / gene annotation 则是后分析能否无缝转入 GO enrichment 的基础。也就是说,这一页不仅服务分析,还提前塑造了解释层。
当 FASTA 从标准 canonical proteome 扩展到 non-canonical、immunopeptidomics、开放修饰或更宽的 peptide 空间时,数据库本身就开始改变 false discovery pressure。数据库被怎样拆组、怎样命名、怎样写入 report,会直接影响 per-group precursor FDR、protein inference 和后续蛋白层交付边界。
Entrapment 研究与 HUPO poster 都说明,数据库结构本身就可能引入偏置。只要 FASTA 里混入过度共享 MS1 特征的 entrapment 序列,eFDR 就会被放大或扭曲;只要 canonical / non-canonical 或不同来源子库没有被明确拆开,复杂数据库里的风险也会被平均化掩盖。
因此 Databases Perspective 里真正需要固定的是:FASTA 版本、分组逻辑、parsing rule、cleavage rules 和 custom modifications。它们共同决定 search space 的边界,也共同决定 FDR 该怎样被验证和解释。
Workflow Templates
Settings Perspective 把“分析参数”抽象成可以保存、继承、设置为默认、导出和覆盖的 schema。也就是说,Spectronaut 其实内建了一个工作流模板系统。
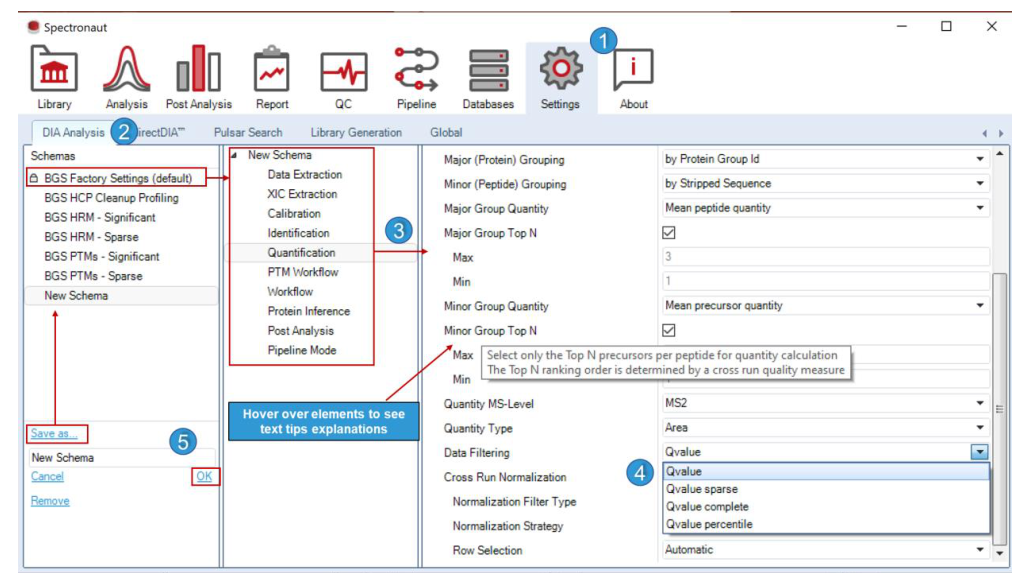
Save as... 让团队把常用 discovery、PTM、快筛、大队列模板沉淀下来;Set as default 则确保后续新项目会自动调用同一套逻辑。这两步加起来,本质上是在防止“每个人都从默认模板胡乱改一遍”。
团队在实际工作中既需要 GUI 可读模板,也需要命令行与版本控制友好的文本格式。JSON override 可在稳定主模板之上覆盖少量参数,既保留统一 SOP,又允许流水线做项目级微调。
蛋白层结果从来不是 precursor 层结果的简单相加。只要 protein grouping、protein inference、single-hit protein 保留策略发生变化,蛋白层的 q-value、蛋白数和经验错误边界就会一起变化。也正因为如此,这些设置不应在项目末尾临时改,而应作为 schema 的正式组成部分固定下来。
对于 discovery 交付,默认的 stratified single hit protein FDR 通常作为保守起点;如果项目确实需要放宽 single-hit protein 处理,应同步转入 entrapment 或经验验证框架,而不是只根据蛋白数变化做决定。
Save as...、Set as default 和导出到 .prop / .txt / .json。这说明 schema 被视为一等对象,而不是附属文件。Tolerance Parameters
质谱搜索和谱图匹配的容差本质上是在 m/z 轴上定义允许误差窗口。Spectronaut 使用 ppm 与 Th,是因为质量分析器检测的是带电离子的 mass-to-charge ratio,而不是肽段的中性分子量。
质谱仪首先把分子转化为带电离子,然后根据离子的 m/z 进行分离、选择、检测和谱图重建。Orbitrap 等傅里叶变换类质量分析器检测离子运动产生的频率信号,再换算为 m/z;TOF 由飞行时间换算 m/z,四极杆按稳定轨迹筛选特定 m/z,离子阱通过捕获、激发或弹出过程获得 m/z 信息。因此,峰匹配时真正比较的是“理论 m/z 与观测 m/z 的差距”。
Da 是质量单位,适合描述中性分子或离子的质量差;但同一段肽在不同电荷态下,同样的质量误差会投影成不同的 m/z 偏移。例如 0.02 Da 的质量误差在 z=1 时约等于 0.02 Th,在 z=2 时约等于 0.01 Th,在 z=4 时约等于 0.005 Th。直接输入 Da 会让软件必须额外推断电荷态和加合离子关系,反而不如直接在 m/z 轴上使用 Th 或 ppm 明确。
该图仅使用演示数值说明单位关系,不代表任何仪器类型或项目的推荐参数。Spectronaut 中,Relative 通常对应 ppm 这类相对误差窗口;Static 通常对应 Th 这类固定 m/z 宽度窗口。Dynamic calibration search 用于评估或校准数据中的质量误差分布;主搜索窗口仍应结合校准后结果、仪器类型与实际 schema 设置确认。
下方默认值为单位换算演示值,不作为 Spectronaut 参数推荐。实际项目应依据校准后的 precursor / fragment mass error 分布确认。
| m/z | 当前 ppm 对应 Th | 当前 Th 对应 ppm |
|---|
ppm 是相对误差;Th 是 m/z 轴上的绝对窗口。相同 ppm 下,m/z 越高,允许的 Th 偏移越大。相同 Th 下,m/z 越高,换算成 ppm 越小。
搜索软件比较的是峰在 m/z 轴上的位置。若中性肽段质量差为 0.02 Da,并且电荷态和加合形式不变,则 z=1、z=2、z=4 分别对应约 0.0200、0.0100、0.0050 Th。直接输入 Da 会把“质量差”与“谱图横坐标窗口”混在一起,容易在不同电荷态之间产生不一致的筛选标准。
可靠的设置不是固定记忆某一个数值,而是检查校准后 precursor / fragment mass error 是否以 0 为中心、分布是否足够窄、是否存在 run 级别漂移。若误差分布明显偏斜,应先排查校准、库来源、raw 文件映射和仪器状态;只有确认系统因素后,才考虑调整容差窗口。
窄且居中:适合较严格窗口;宽或偏移:先检查校准与数据匹配。
ppm 表达的是相对误差:ppm = Δm/z ÷ m/z × 10⁶,反向换算为 Δm/z = m/z × ppm ÷ 10⁶。同样 ±10 ppm,在 m/z 400 处是 ±0.004 Th,在 m/z 1000 处是 ±0.010 Th。对于 Orbitrap、TOF 等高分辨数据,ppm 更容易让低 m/z 与高 m/z 区间保持相近的相对质量精度标准。
Thomson 是 m/z 单位,常用于表达固定 m/z 容差窗口。±0.02 Th 表示理论峰左右各允许 0.02 m/z。固定 Th 的优点是直观;风险是对低 m/z 可能过宽,对高 m/z 可能过窄。低分辨 MS/MS 或宽校准窗口中常见固定 m/z 窗口,高分辨主搜索通常更偏向 ppm。
Da 描述质量差,不直接描述谱图横坐标。在电荷态和加合形式不变时,中性肽段质量差投影到 m/z 轴上的偏移约为 ΔTh = ΔDa ÷ z。因此同一个 Da 误差在 z=1、z=2、z=3 的 m/z 表现不同。Spectronaut 让用户设置 Th 或 ppm,是为了让搜索窗口直接落在仪器实际检测坐标上。
容差过窄会丢失真实峰,表现为鉴定率下降、碎片匹配减少或 run 间一致性变差;容差过宽会引入更多候选峰和干扰峰,增加 FDR 控制和定量边界判断压力。稳定项目应结合校准后 mass error 分布、仪器类型、MS1/MS2 分辨率和项目目标确定容差,而不是只追求更小数值。
Global Settings
这四页控制的不是单次实验,而是软件在整个电脑或工作站上的日常行为。
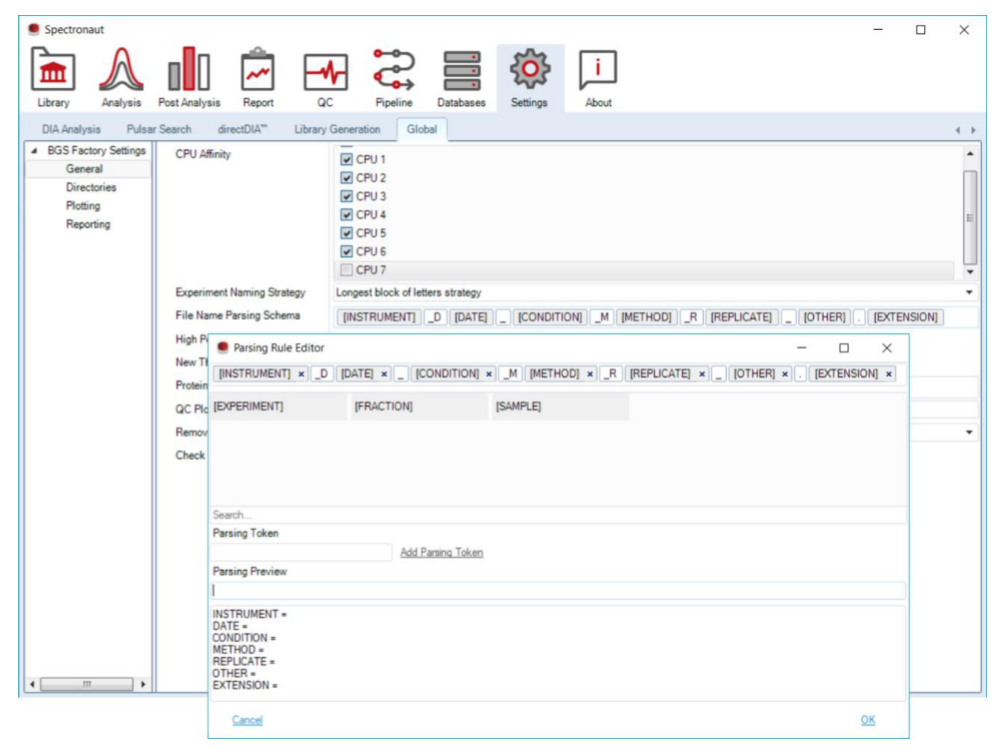
File Name Parsing Strategy 被放在 General 里,是因为它实际上决定了软件能否从文件名中自动读出 condition、replicate、fraction、批次等信息。对于大队列或多用户环境,这一步是减少人工注释失误的最大杠杆。
Directories 管的不只是输出目录,还包括 Search Archive、临时文件和结果保留位置。本地高速盘用于承接高频读写,路径规划本身就是性能管理的一部分,而不是办公整理习惯。
Plotting 里常见的积分边界、预期洗脱时间和曲线平滑设置,并不会改变底层信号,但会显著改变你如何理解峰形、如何判断边界、如何做人工 review。因此它直接影响读图体验和判断一致性。
Global Reporting 决定 Pipeline 结果放哪、文件怎么命名以及哪些输出被默认保留。它应与交付规范、命名规则、结果归档目录一起统一,不然不同人导出的产物会越来越难管理。
General 负责 software-level defaults,其中 file name parsing schema 是自动注释的核心;这一步一旦统一,后面的 Condition Setup 能省去大量手工输入。Directories 负责 Search Archive、managed FASTA、libraries、results、temp 等默认路径。前面关于本地高速盘的要求,会在这一层真正落到可执行设置上。Plotting 会改变 review 时你看到的图像样式与边界判读逻辑,例如 expected elution time、XIC integration display 和 smoothing 相关选项,因此它直接影响人工复核的一致性。Reporting 则把自动输出、命名与结果保留策略统一到软件层,让 pipeline 不至于每个项目都长出不同的产物结构。Command Line
Spectronaut Command Line Mode 覆盖许可证、建库、DIA 分析、directDIA、结果合并、SNE 文件管理、全局环境选项和退出码。它与 GUI 使用同一套 settings schema、report schema、FASTA、GO annotation 和目录设置,因此适合把已经验证过的图形界面流程迁移到批处理、调度器和平台化 SOP。
Windows 可在 cmd 或 PowerShell 中运行,Linux 可在终端中运行。实际可执行入口依安装方式而定,Windows 常见形式为 dotnet Spectronaut.dll,Linux 常见形式为 spectronaut。查看总帮助使用 spectronaut -h,查看某个子命令使用 spectronaut <command> -h。
spectronaut activate <license_key> 用于命令行激活许可证。spectronaut deactivate 用于释放当前机器上的许可证。sudo dpkg -i spectronaut-VER.deb 安装指定版本;其他 Linux 发行版按 tarball installer 中的 Read me 执行。lg、diaanalysis、direct、convert、combine 和 manageSNE。lg 负责 library generation;diaanalysis 使用预编译 library 或 search archive 做 DIA 分析;direct 执行 library-free directDIA;convert 把 vendor raw 转为 HTRMS;combine 执行 SNE Combine 流程;manageSNE 用于加载、导出、合并、解锁或检查 SNE 文件。
命令行不是另一套分析逻辑,而是把 GUI 中已经配置好的 schema、conditions、report 和目录规则变成可重复调用的文本接口。
使用 Pulsar 建库时,命令从 spectronaut lg -se Pulsar 开始。后续参数定义输入 raw 文件、Search Archive、Managed FASTA、搜索 schema、library generation schema、Search Archive 输出、library 输出、实验名、protein inference FASTA 和 GO annotation。
| 参数 | 功能 | 使用要点 |
|---|---|---|
-se Pulsar | 指定使用 Pulsar 执行 library generation。 | 作为 Pulsar 建库流程的第一个关键参数。 |
-r [file] | 加入单个 raw 文件,支持 GUI 中可识别的 vendor 格式。 | 可重复使用,用于逐个追加文件。 |
-d [directory] | 加入目录内所有 Spectronaut 可识别 raw 文件。 | 可重复使用;包含 Bruker .d、Waters run folder 等目录型 vendor 数据。 |
-sa [file] | 加入指定 Search Archive *.psar。 | 可重复使用;默认目录由 Global Directories 管理。 |
-sad [directory] | 加入目录中的全部 Search Archives。 | 适合批量合并历史搜索记录或项目内多来源 archive。 |
-fasta [file] | 指定用于 DDA 搜索空间的 Managed FASTA *.bgsfasta。 | Managed FASTA 包含 header parsing rule;可重复使用。 |
-rs [schema] | 指定 Pulsar Search schema。 | 可传 *.prop 路径,也可传 GUI repository 中已有 schema 名称。 |
-es [schema] | 指定 Library Generation schema。 | 未指定时使用默认 schema。 |
-a [file] | 指定本次搜索生成 Search Archive 的输出位置。 | 未指定时写入默认 Search Archive 目录。 |
-k [file] | 指定生成 spectral library 的输出位置。 | 未指定时写入默认 Spectral Libraries 目录。 |
-regex [pattern] | 对 -d 指定目录应用正则过滤。 | 适合只选择特定批次、后缀或命名规则的文件。 |
-n [text] | 设置实验名。 | 会用于标记 resulting library 和 Search Archive。 |
-inf [file] | 指定 protein inference 使用的 Managed FASTA。 | 默认使用搜索空间 FASTA;需要独立 inference FASTA 时显式指定。 |
-go [file] | 加入 Gene Annotation 文件。 | 可重复使用,服务于后续 GO 相关结果解释。 |
外部搜索引擎建库从 lg -se <SearchEngineName> 开始,支持 ProteomeDiscoverer、MaxQuant、ProteinPilot、BGSGenericSearchFormat 和 Mascot。这一流程的重点是把搜索结果、对应 raw 文件、输出 library、library generation schema 和 protein inference FASTA 对齐。
| 参数 | 功能 | 使用要点 |
|---|---|---|
-se <SearchEngineName> | 选择外部搜索引擎结果类型。 | 名称必须与支持列表一致。 |
-sr [search results] | 指定外部搜索结果路径。 | 不同搜索引擎需要的结果文件类型不同,必须与对应格式匹配。 |
-rd [raw files] | 指定 run files 路径。 | 用于把搜索结果与原始采集数据关联起来。 |
-o [library file] | 指定输出 library 文件,包含文件名。 | 这是外部结果转库流程的主要产物。 |
-s [schema] | 指定 Library Generation schema。 | 通常传 *.prop 路径;未指定时使用默认 schema。 |
-fasta [file] | 指定用于 protein inference 的 Managed FASTA。 | 用于保证蛋白归并和数据库解析规则一致。 |
spectronaut diaanalysis 用于带预编译 library 或 Search Archive 的 DIA 分析;spectronaut direct 用于 library-free directDIA。两者共享大量输入、schema、输出、FASTA、GO、condition 和 pipeline integration 参数。
| 参数 | 功能 | 使用要点 |
|---|---|---|
-h | 显示当前命令帮助。 | 用于确认当前版本可用参数。 |
-r [file] | 加入单个 run file。 | 支持 *.htrms、*.raw、*.wiff、*.bgms、_HEADER.TXT、analysis.baf 等。 |
-d [directory] | 加入目录内全部可识别 run files。 | 目录型 vendor 文件也会被识别。 |
-regex [pattern] | 对目录输入应用正则过滤。 | 常用于批量目录中只选择部分 runs。 |
-a [library] | 把同一 spectral library 分配给实验中的每个 run。 | 可重复使用,适合多 library 输入。 |
-ar [library] | 把 spectral library 分配给最后一次 -r 加入的 run。 | 适合 run 与 library 一一匹配的复杂输入。 |
-s [schema] | 指定 DIA Analysis settings schema。 | 可传 *.prop 或默认 schema repository 中已有名称。 |
-rs [schema] | 指定 report schema。 | 决定自动导出字段和报告结构。 |
-o [directory] | 指定输出目录。 | 结果会写入包含分析日期和实验名的子目录;Windows 未指定时使用默认结果目录,Linux 未指定时使用当前目录。 |
-n [text] | 设置实验名。 | 未指定时由 run file names 自动生成。 |
-fasta [file] | 指定 FASTA 或 Managed FASTA。 | library-based 分析中用于 protein inference;directDIA 流程中为必需输入。 |
-go [file] | 加入 GO annotation 文件。 | 可重复使用,便于后续注释与富集解释。 |
-con [file] | 导入 condition setup。 | 推荐从 GUI 导出;未指定时会按 run name parsing 推导 condition names。 |
-command [file] | 读取 command arguments file。 | 必须作为第一个参数;后续命令行参数会被忽略。该文件可由 experiment setup wizard 最后一页生成。 |
-rExt [file] | 为 1-step hybrid library-based DIA analysis 加入单个 extension run。 | 支持与常规 run 相同的 vendor 文件类型。 |
-dExt [directory] | 为 1-step hybrid library-based DIA analysis 加入目录内 extension runs。 | 适合 hybrid library 扩展数据集中管理。 |
-sne [path] | 加载 .sne experiment。 | 该方式已弃用,SNE 文件处理优先使用 manageSNE。 |
-j [json] | 使用 JSON 文件做 settings override。 | 用于固定主 schema 之上的项目级参数覆盖和 flexible pipeline integration。 |
--inputNormalizationSNE [file] | 为 PTM Stoichiometry calculation 加入 input normalization reference SNE。 | 仅在启用 PTM Localization 设置时生效。 |
--sampleLinking [file] | 加入样本 linking 文件。 | 可由启用 PTM Localization 的 Experiment setup wizard 生成。 |
示例结构:dotnet Spectronaut.dll -d "C:\data\My Experiment" -a "C:\data\My Experiment\library.txt" -s "my_settings" -o "C:\data\My Experiment\Results" -n "My Experiment 1" -f ".*\.wiff"。如果 library 自动解析失败,应先在 GUI 中加载 library 并确认必要列被正确识别,再迁移到命令行。
spectronaut combine 执行 SNE Combine pipeline。它适合从多个 .sne 文件生成统一输出,而不是把所有原始 runs 重新放进同一个超大实验里。
| 参数 | 功能 | 使用要点 |
|---|---|---|
-s [settings schema] | 选择 combine 使用的 settings schema。 | 用于固定 combine 后处理规则。 |
-n [name] | 指定实验名。 | 用于输出命名和结果归档。 |
-fasta [file] | 指定 protein inference 使用的 FASTA 或 Managed FASTA。 | 确保合并结果的 protein grouping 规则一致。 |
-sne [file] | 指定要 combine 的 .sne 文件。 | 可重复使用。 |
-d [directory] | 加入目录中的所有 .sne 文件。 | 可重复使用,适合批量结果目录。 |
-o [directory] | 指定输出目录。 | 必需参数。 |
-rs [report schema] | 指定 custom report schema。 | 决定输出报表字段。 |
spectronaut manageSNE 面向已经存在的 .sne 文件。它可加载实验生成报告,也可用 --merge 在 experiment level 合并多个 SNE,生成等同于一起运行的结果。大型实验使用 --merge 可能消耗大量磁盘和内存;仅生成组合报告时优先考虑 combine。
| 参数 | 功能 | 使用要点 |
|---|---|---|
-sne [file] | 选择要加载、处理或合并的 SNE 文件。 | 可重复使用。 |
-d [directory] | 加载目录中的所有 SNE 文件。 | 适合批量处理。 |
-n [name] | 指定实验名。 | 用于输出和新实验命名。 |
-o [directory] | 指定输出目录。 | 必需参数。 |
-rs [report schema] | 指定 custom report schema。 | 用于从 SNE 导出指定字段结构。 |
--merge | 当指定多个 SNE 时,把它们在 experiment level 合并为一个新的 Spectronaut experiment。 | 可能超过可用磁盘或 RAM;大型项目要谨慎。 |
--skip-loading | 不加载和处理 experiment。 | 可用于快速版本兼容性检查或解锁文件。 |
--unlock | 解锁由 Bruker ProteoScape 环境创建的 SNE,便于共享。 | 批量解锁时可与 --skip-loading 联用。 |
部分选项可不带分析子命令单独执行,也可放在子命令前生效。例如 dotnet Spectronaut.dll -setTemp <path_to_temp> direct -d <raw_path> -fasta <fasta_path> -o <output_path> 会先设置临时目录,再执行 directDIA 分析。
| 参数 | 功能 | 影响范围 |
|---|---|---|
-setTemp <directory> | 指定 Spectronaut temporary directory。 | 影响运行时临时文件位置,通常与高速本地盘规划相关。 |
--version | 输出 Spectronaut 版本并退出。 | 用于脚本记录版本或兼容性检查。 |
--exportModRepository <file> | 导出 modification repository。 | 用于迁移或版本化自定义修饰库。 |
--importModRepository <file> | 导入 modification repository。 | 会覆盖当前 repository,执行前需确认版本。 |
--exportEnzymeDB <file> | 导出 enzyme database。 | 用于固定 cleavage rules 的团队版本。 |
--importEnzymeDB <file> | 导入 enzyme database。 | 冲突 enzyme names 会被覆盖。 |
--noXICDump | 关闭 automatic XIC dump。 | 会持续生效,直到用 XIC export directory 选项重新开启。 |
--setXICExportDirectory <dir> | 开启 automatic storing of all XICs as SQL database files。 | 用于把 XIC 证据接口固定到指定目录。 |
退出码用于外部脚本判断任务状态。0 表示成功;1-9 为 activation related,例如 activation error、deactivation error、insufficient license、软件版本或 activity 不匹配;11-19 为 setup errors,例如 command-line invocation 不正确;>=20 为 later-stage errors,表示分析后续阶段发生错误。调度系统应根据这些分组决定停止、重试、报警或继续后续任务。
在 GUI 的 experiment setup wizard 最后一页可生成示例 arguments file。命令行使用 -command [file] 读取该文件时,必须把 -command 放在第一个参数位置,后面的普通命令行参数会被忽略。这个机制适合把 GUI 验证过的配置保存成文本,再交给脚本或调度器执行。
-j [json] 面向 flexible pipeline integration。主 settings schema 负责稳定继承,JSON override 负责局部覆盖项目差异,适合自动化系统在不复制大量 schema 文件的情况下调整少量参数。
Integration Layer
HTRMS Converter 是与 Spectronaut 一起安装的配套转换工具,用于把 DIA run files 转成 Biognosys 兼容的 HTRMS 格式。HTRMS 文件已经完成预处理并为 Spectronaut 分析优化,适合同一批 raw 数据需要重复分析、反复调参或进入自动化流水线的场景。BGMS Raw API 则提供更底层的 C# .NET 接口,用于构建自定义 raw vendor processor。
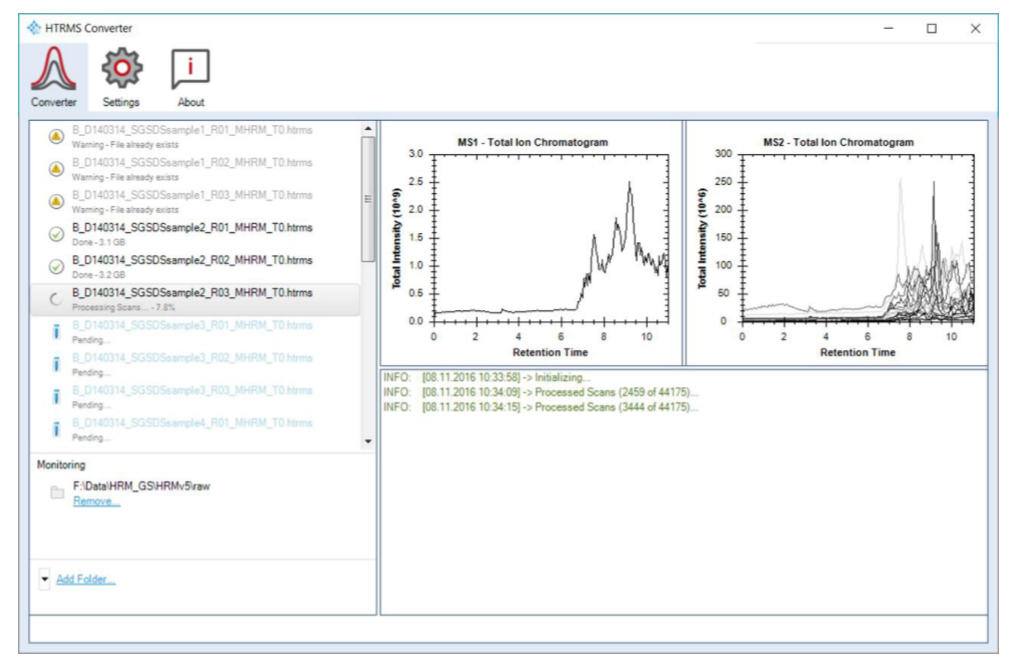
HTRMS 是 Biognosys 兼容格式,转换后的文件可减少后续重复分析时间。Converter 不需要 Spectronaut license key,可部署在多台电脑上用于前置转换,因此适合把 raw 文件预处理从正式分析节点中拆分出来。
在 Converter perspective 中选择 Add Files... 后,可一次选择一个或多个 MS/MS 文件。确认 conversion parameters 后,任务会加入主 task list;之后还可以继续追加任务。针对 overlapping windows 采集的数据,可在 HTRMS file settings 中启用 MS2 Demultiplexing,对 varied-size 或 fixed-size MS/MS scans 进行解卷积处理。
Add Folder... 会指定输入目录,并自动转换目录中符合基本过滤条件的有效 MS/MS 文件。Folder Conversion 提供 Batch Conversion settings,可按 vendor、file age 等条件过滤,也可以监控文件夹,使新增 raw 文件被自动转换。
BGMS 是 Biognosys generic MS file format。BGSRawAPI.dll 随 Spectronaut 一起安装,基于 C# .NET,可用于构建自定义 raw vendor processor。典型场景包括仪器格式尚未被 Spectronaut 原生支持,或 DIA 方法需要特殊 scan preprocessing。
Windows 可直接调用 HTRMSConverter.exe -i [PATH] -o [PATH] -s [mySettings] -nogui。也可以通过 Spectronaut 的 converter 入口在 Windows 或 Linux 上调用转换任务。转换命令的参数很少,但每个参数都直接决定输入来源、输出位置、转换 schema 和是否使用 GUI。
| 参数 | 是否必需 | 功能 | 默认逻辑 |
|---|---|---|---|
-i [path] | TRUE | 指定 raw file,或包含 raw files 的目录。 | 无默认值,必须提供。 |
-o [path] | FALSE | 指定 HTRMS 输出文件路径,或输出文件夹。 | 默认写在 raw 路径旁,并把文件后缀替换为 .htrms。 |
-s [schema] | FALSE | 指定 conversion settings schema 的路径或名称。 | 默认使用 BGS_FactorySettings。 |
-nogui | FALSE | 在命令行窗口中运行转换,而不是启动 UI task。 | 未提供时按界面任务方式启动。 |
Operational SOP
这一部分把配置管理压成实验室级规范,直接对应实际平台管理中的执行链。
先统一 FASTA 版本、GO 注释、自定义修饰和 cleavage rules,再允许建库和分析启动。这样后续项目之间才有横向可比性。
至少区分 discovery、PTM、快筛和大队列四类 settings schema,并明确哪些可以改、哪些只能继承。
把 Search Archive、temp、reports、plots 和 XIC 导出目录全部放进标准结构里,同时约定 file-name parsing 规则,减少人工注释。
先在 GUI 内跑通并导出 arguments file,再迁移到命令行。这样能最大限度减少“命令写错”和“逻辑对不上”的情况。
对原始文件转换、结果留存、Exit Codes 和失败重试做统一规定,软件才算真正进入平台运行状态。
Further User Resources
这一组资源直接服务数据库管理、Linux pipeline、directDIA 参数覆盖、Skyline library 导出和软件环境合规说明。
该 FASTA 只包含 iRT kit 相关融合蛋白序列,用于在数据库层明确 iRT peptide 的来源。正式项目中,iRT 的作用不是增加蛋白发现深度,而是建立 retention time 校准和长期 QC 的坐标系。
>Biognosys|iRT-Kit_WR_fusion LGGNEQVTRYILAGVENSKGTFIIDPGGVIRGTFIIDPAAVIRGAGSSEPVTGLDAKTPVISGGPYEYRVEATFGVDESNAKTPVITGAPYEYRDGLDAASYYAPVRADVTPADFSEWSKLFLQFGAQGSPFLK
第三方依赖清单说明 Spectronaut 的平台能力由多个层面组成:Thermo、Bruker、SCIEX、Waters 等 raw data reader 负责仪器文件读取;OpenXml、PDFsharp、ZedGraph 和 SQLite 支撑报表、图形和本地数据库;Json.NET、System.CommandLine、Microsoft.ML、LightGBM 与 ONNX Runtime 支撑序列化、命令行和机器学习组件。
在进行软件环境确认或 IT 合规审查时,可重点核对 raw data reader、命令行组件、机器学习运行库、本地数据库和报表依赖是否满足项目要求。
convertFASTA 用于把 plain-text FASTA 转换为 Managed FASTA .bgsfasta。自定义 header parsing rule JSON 通过 --parsingRule <rule.json> 传入,只在 FASTA 转换阶段使用;directDIA settings JSON 则通过分析命令的 -j 使用,两者不能混用。
dotnet Spectronaut.dll convertFASTA -fasta "D:\db\protein.fasta" --parsingRule "D:\rules\custom-rule.json" -o "D:\managed-fasta"
规则 JSON 的核心字段为 name、proteinIDToken、keywords 和 parsingRule。其中 parsingRule 用固定文本和 $[Token]$ 组合定义 header 如何被拆成 accession、gene、organism 和 description 等字段。
命令中 -fasta 与 -o 是必需参数;-setTemp 可把临时目录放到本地高速盘;-v 用于输出更详细的错误;--terminateAfterError 可在批处理前强制遇错即停。
directDIA JSON settings 用于命令行 -j 参数覆盖默认设置或 -s 指定的 schema。结构上分为 DIA_Analysis 与 Pulsar_Search:前者控制定量、FDR、归一化、imputation、PTM localization 和 XIC extraction;后者控制酶切规则、修饰、搜索模式、directDIA workflow、质量容差和离子类型。
进入批处理前,应重点检查 q-value / PEP 是否在 0 到 1 之间、single-hit protein 规则是否明确、肽段长度和 missed cleavages 是否为非负整数、DirectDIA_Workflow 是否为 directdia_deep、directdia_fast 或 directdia。
相关专题
Analysis 与 Reporting 分别承接 workflow 设计、schema、QC、pipeline 和 XIC 接口。