保留碎片离子证据
蛋白质组定量从碎片离子强度推断肽段和蛋白强度。过早过滤会丢掉弱但真实的证据。

Missing Values
在单细胞蛋白质组矩阵中,一个 NA 可能代表真实低表达,也可能代表真实存在的弱信号被算法过滤。后者如果不被识别,会直接影响聚类、差异分析和通路解释。

单细胞组学分析通常从一张矩阵开始:行是细胞,列是分子特征。转录组里是基因表达矩阵,蛋白质组里则是蛋白质定量矩阵。如果某个蛋白在一部分细胞中有数值、在另一部分细胞中没有数值,矩阵就出现了缺失值。
这个空格可能代表真实生物学差异,也可能只是输入量太低、信噪比不足或局部打分略低导致的算法性缺失。对单细胞蛋白质组来说,这个区别尤其重要:研究者关心的正是细胞之间的差异,如果缺失值主要来自算法不稳定,就可能把“检测失败”误读为“细胞异质性”。
因此,本页的主线不是比较不同质谱类型,而是解释 Full-DIA 如何把缺失值问题前移,在原始信号解析和全局错误率控制阶段减少缺失。相关背景可交叉阅读 单细胞蛋白组专题、DIA 基础 和 FDR 控制。
From Imputation To Prevention
传统做法是先得到稀疏矩阵,再用统计模型填补;Full-DIA 的思路是从源头减少缺失,在鉴定、定量和错误率控制过程中保留更多可信证据。
蛋白质组定量从碎片离子强度推断肽段和蛋白强度。过早过滤会丢掉弱但真实的证据。
如果同一肽段在多数样本中稳定出现,少数样本的边缘缺失就值得回到原始信号重新检查。
减少缺失值不能以增加假阳性为代价,最终矩阵必须在实验层面接受 FDR 约束。
深度学习模型可利用碎片离子结构、共洗脱质量和跨样本模式,对低置信度强度进行去噪。
Algorithm
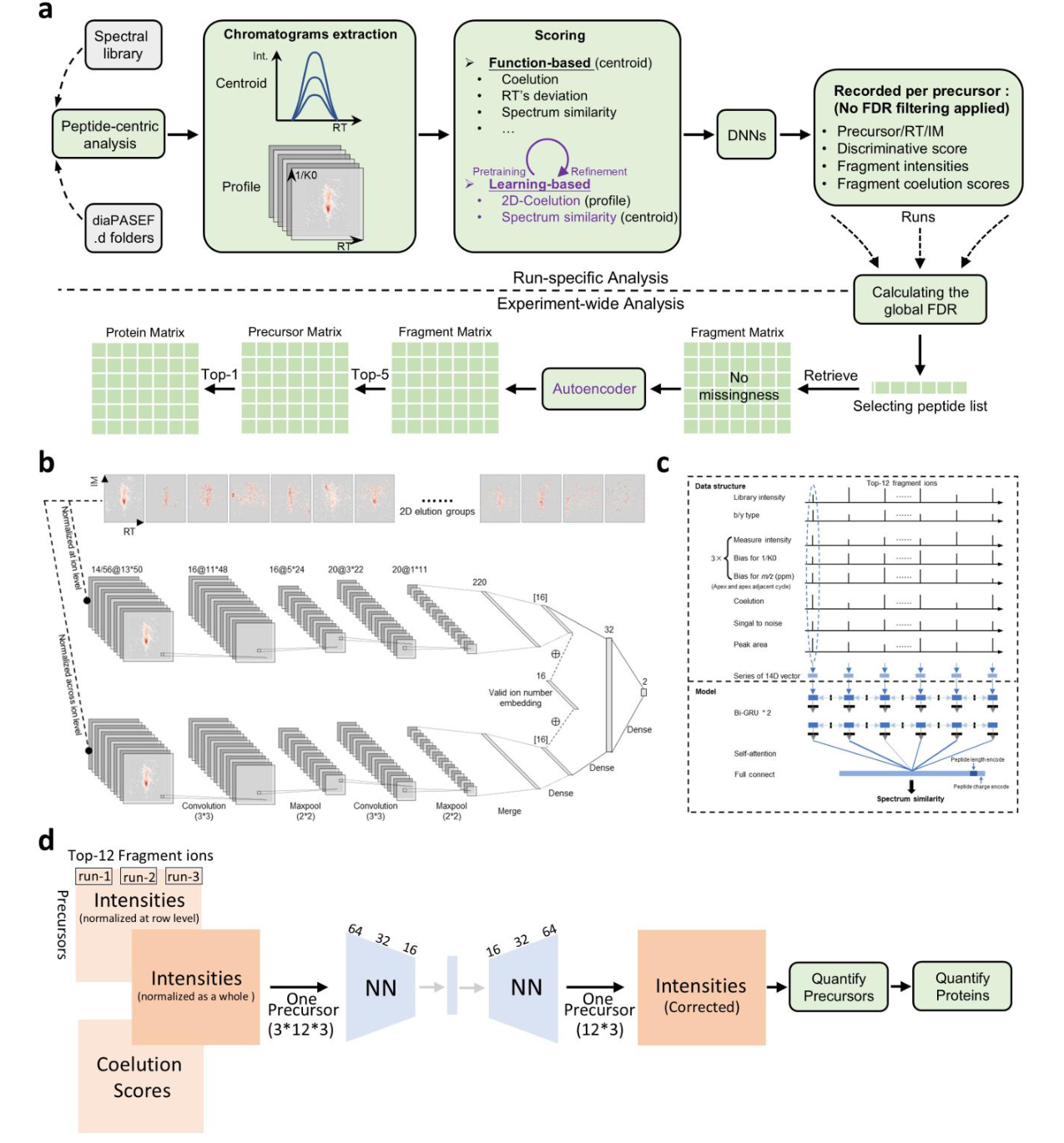
Full-DIA 的流程可以理解为三步:准确找到真实肽段、把碎片离子强度组织成完整矩阵、用深度学习校正低质量测量。
| 模块 | 解决的问题 | 对应技术原理 |
|---|---|---|
| Peptide-centric analysis | 从谱图库目标肽段出发,在每个样本中寻找对应洗脱信号。 | DIA 数据解析 |
| DeepProfile | 学习多离子共洗脱结构,判断弱信号是否形成可信峰组。 | AI 与深度学习打分 |
| DeepMall | 比较观测碎片与谱图库预期碎片,同时纳入质量偏差、信号质量和碎片类型。 | 软件鉴定与谱图相似性 |
| Global FDR | 在整个实验层面筛选可信肽段和蛋白,而不是只依赖单个 run 的局部阈值。 | FDR 与错误率控制 |
| DeepQuant | 用自监督自编码器对碎片离子强度进行去噪和校正,生成更可信的定量矩阵。 | 定量矩阵与 QC |
Results
Full-DIA 的优势不是单一指标提升,而是从鉴定、定量、矩阵完整性到生物学解释的一整条链路更稳。
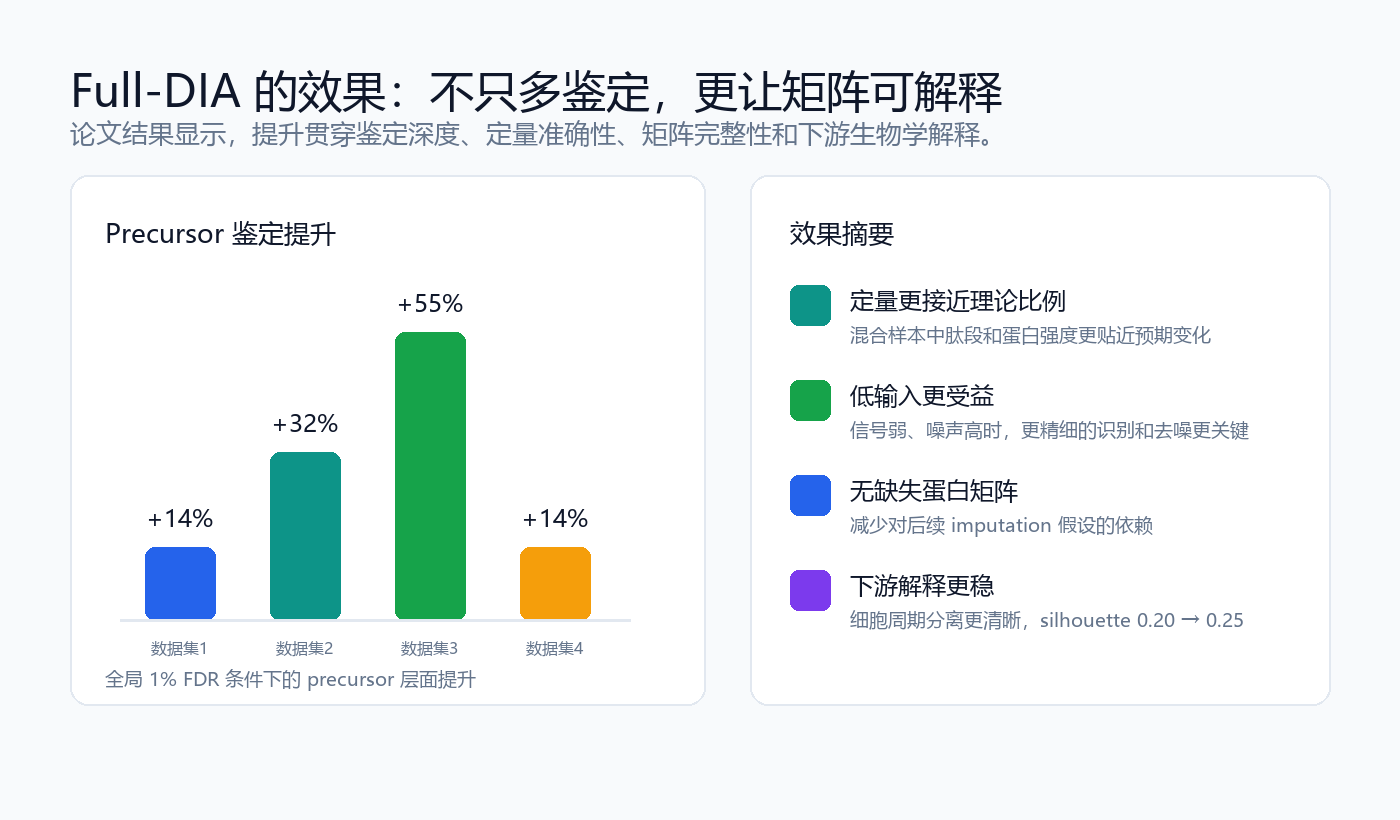
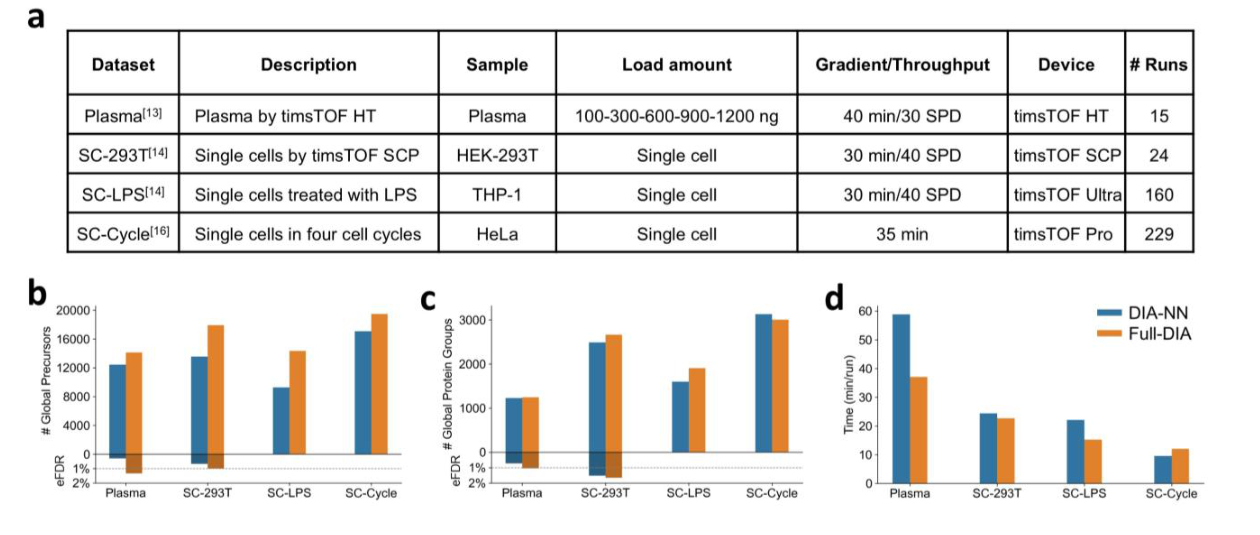
在全局 1% FDR 条件下,Full-DIA 在多个测试数据集上报告了更多 precursor 鉴定。论文中四个数据集的 precursor 层面提升分别为 14%、32%、55% 和 14%;蛋白组层面的变化分别为 1%、7%、19% 和 -4%。这说明 Full-DIA 对肽段和 precursor 层面的弱信号挖掘更稳定,而蛋白层面的收益会受到蛋白归并和共享肽段等因素影响。
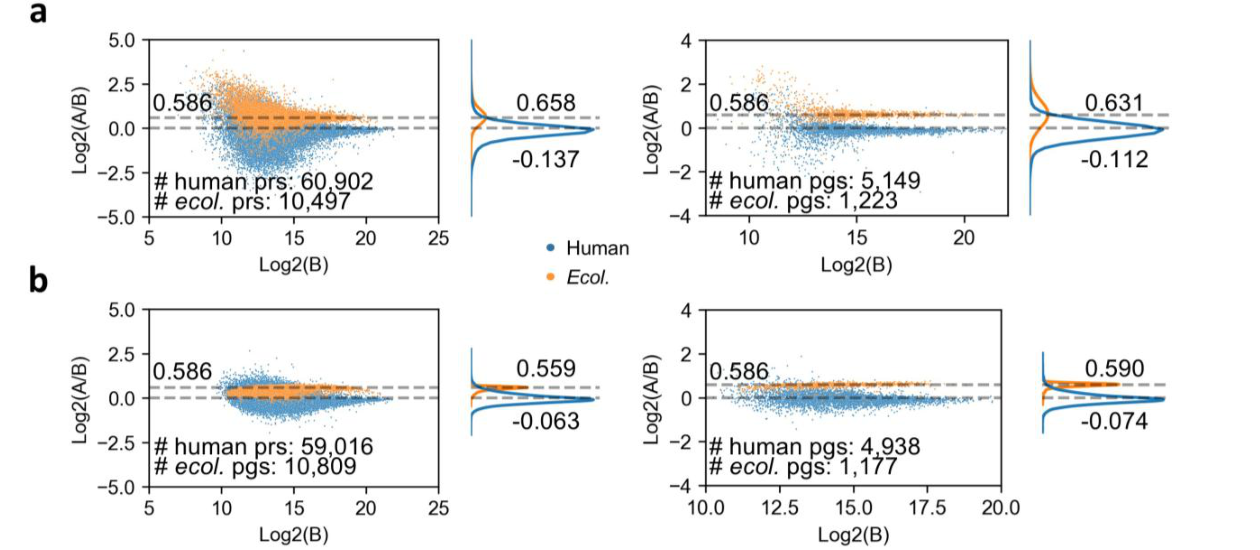
定量准确性同样关键。减少缺失值不能牺牲定量准确性,否则完整矩阵只是“看起来完整”。论文中的已知比例混合样本和梯度样本评估显示,Full-DIA 在肽段和蛋白层面保持或提升了定量准确性,尤其在低输入、弱信号和高噪声场景中更能体现算法优势。
Biological Interpretation
当缺失值减少后,细胞聚类、差异蛋白分析、通路富集和跨条件整合都会更稳,结论更不容易被检测深度和缺失模式带偏。
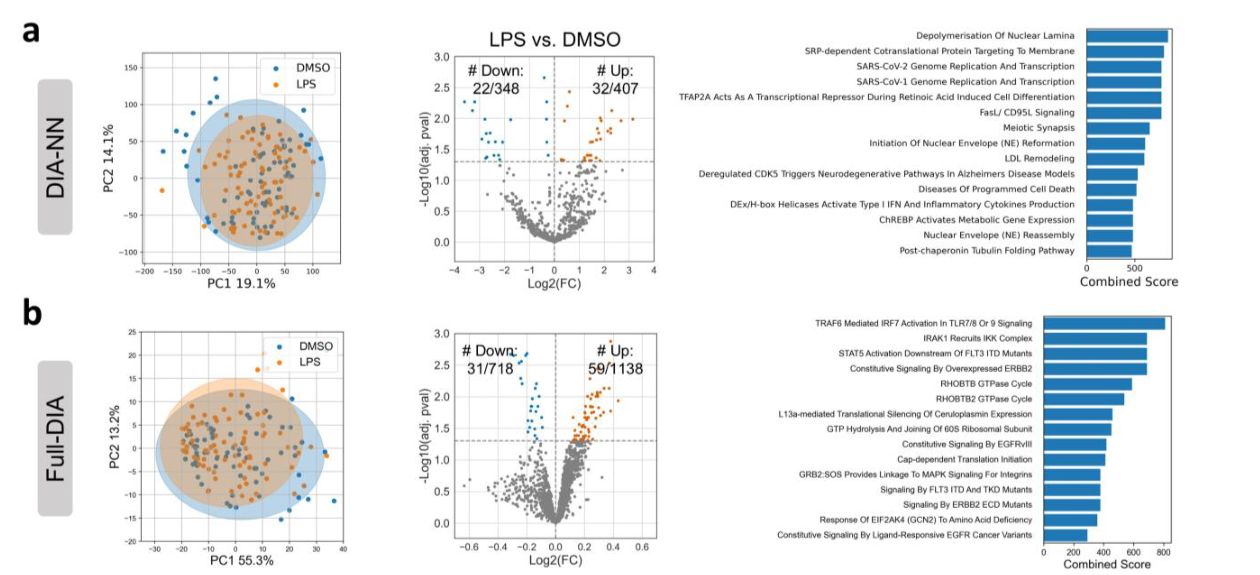
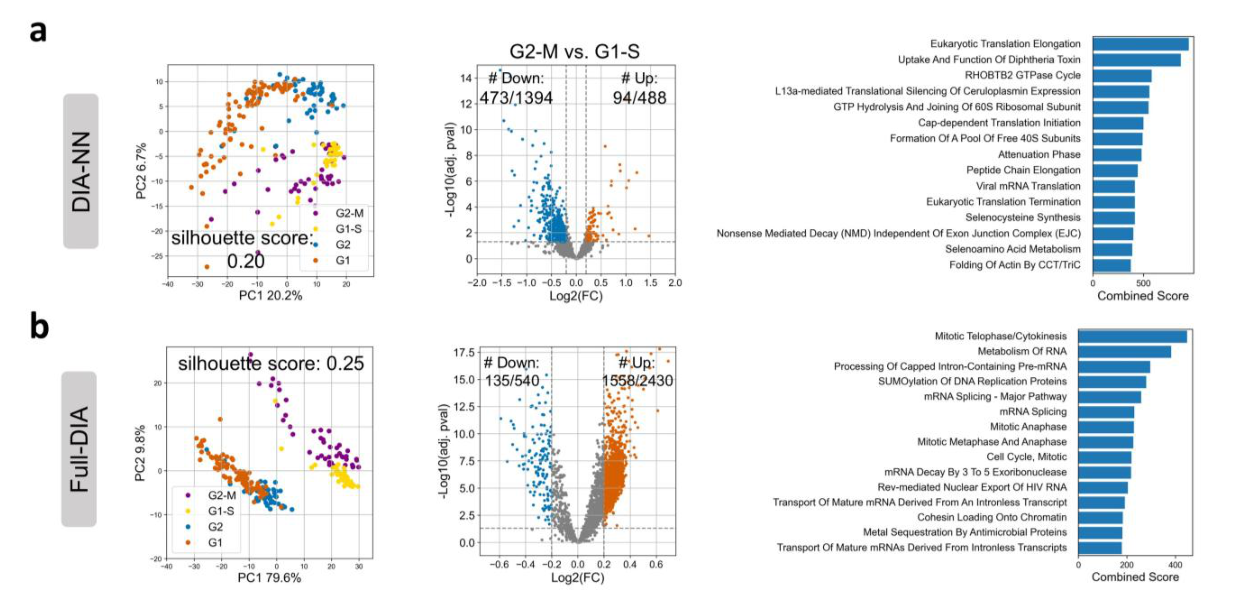
Translation
“从源头减少缺失值”的思想不局限于单细胞。低输入蛋白质组、大规模队列、临床样本和其他多组学矩阵都会面对弱信号、跨样本匹配和缺失值问题。
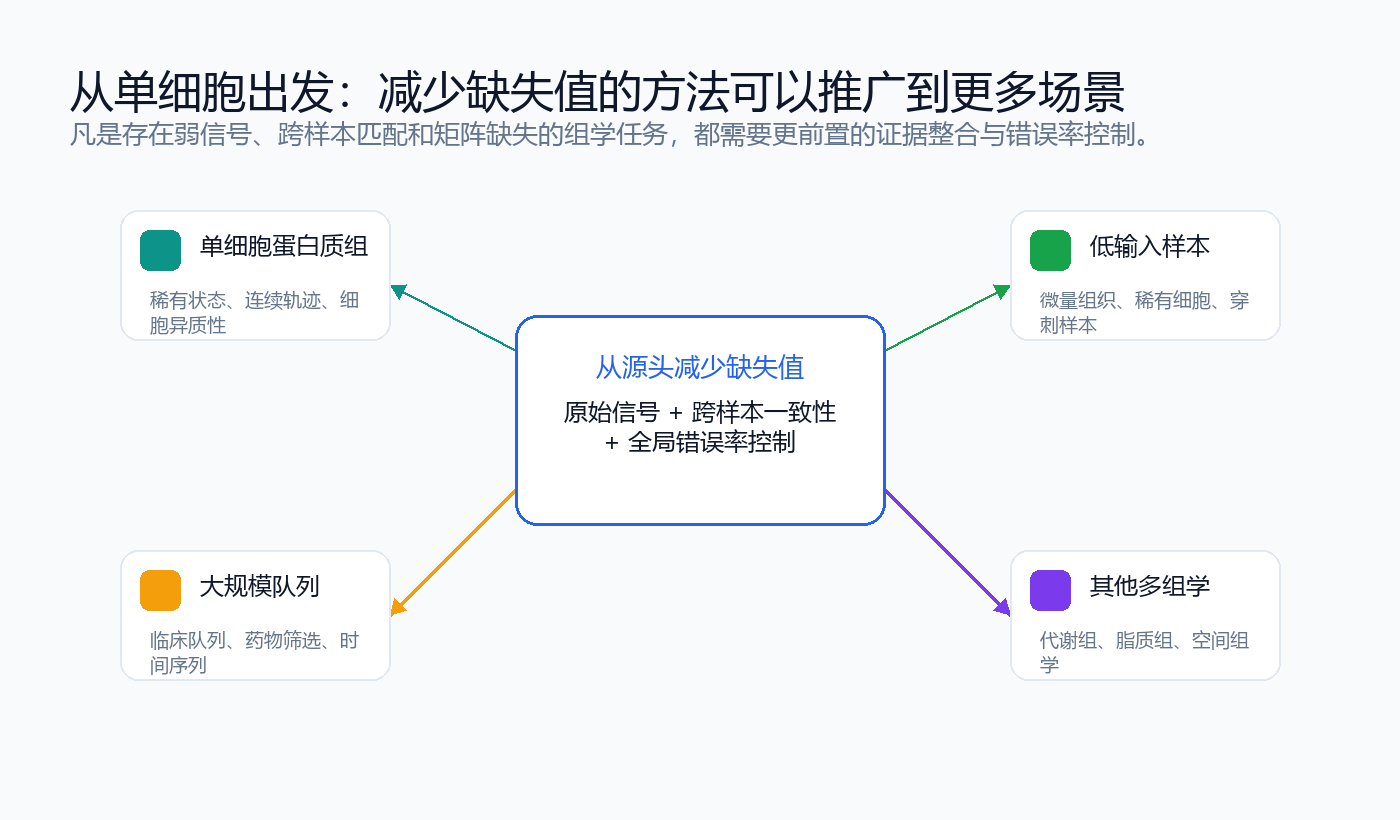
微量组织切片、稀有细胞群、临床穿刺样本和激光显微切割样本都可能受益于更完整的矩阵构建思路。
临床队列、药物筛选和时间序列实验中,队列级证据整合可减少由局部阈值造成的假阴性。
代谢组、脂质组和空间组学也常见弱信号、跨样本匹配和矩阵缺失问题。
矩阵完整性、错误率控制和下游解释应作为项目复核的共同指标,而不是只看鉴定数量。
Cross References
本页可与以下站内技术原理页面配合阅读,形成从方法原理到软件流程再到单细胞解释的闭环。