本章核心内容
Spectronaut 在这一层把“原始数据”组织成“可统计比较的条件矩阵”。如果条件注释、workflow 选择或 settings schema 选错,后面的 q-value、fold change 和 candidates 都会被带偏。
分析流程
Spectronaut Analysis 覆盖 library-based DIA、directDIA、directDIA+、
PTM probing、Method Evaluation、conditions 与
review / refinement,对应 Spectronaut 中最核心的分析、条件注释和复核部分。
总述
Library Perspective 决定你带什么知识进入分析,Post Analysis 决定你怎么解释结果,而 Analysis Perspective 正处在中间,负责把 raw file、FASTA、library、settings 和 conditions 组合成真正可比较的实验对象。
Spectronaut 在这一层把“原始数据”组织成“可统计比较的条件矩阵”。如果条件注释、workflow 选择或 settings schema 选错,后面的 q-value、fold change 和 candidates 都会被带偏。
从 raw、FASTA、library 到 workflow、conditions、review 和交付层的关系图。
工作流矩阵
不同 workflow 并不是同一个按钮的不同皮肤,而是对输入、搜索空间、速度和解释方式的不同承诺。
| 路线 | 输入要求 | 适用场景 | 要避开的误用 |
|---|---|---|---|
| Library-based DIA | DIA raw + 高质量谱图库,可配 FASTA 与 GO | 已有成熟建库体系、目标是稳定大队列分析 | 不要把过时或不匹配的 library 当成万能增益 |
| directDIA | DIA raw + FASTA,可配 GO | 快速启动 discovery、没有现成谱库 | 不要以为省掉 library 就省掉了 settings 和 review |
| directDIA+ Fast / Deep | 与 directDIA 相同 | 需要在速度、覆盖深度和 search space 之间做权衡 | 复杂 PTM search space 下不要只看 Fast 更快 |
| PTM Probing | DIA raw + FASTA + 明确的修饰探索目标 | 修饰不确定、希望开放探索 | 不要把开放探索结果直接当作最终机制结论 |
| Method Evaluation | directDIA workflow + 多种 DIA 方法条件 | 比较不同 acquisition methods | 该模式不适合正式定量实验 |
当 Analysis Settings Schema 被确定时,q-value 阈值、对象层级、protein inference 与 data filtering 就已经进入工作流主干。这一步定义的是本次实验以什么可信度结构进入后续统计。
进入更开放的 search space 后,FDR 不能只看成 settings 里的一个数字。它需要和 precursor 层控制、protein 层解释边界、entrapment 验证和后续报表字段一起联读。
Library-Based DIA
当你已经有稳定的 谱库文件时,这条路线往往能提供更高的覆盖和更稳定的后分析结构,但代价是你必须对 library 的来源、版本和适用性负责。
directDIA
directDIA 被设计成显式向导,说明它并不是黑箱按钮。你依然需要定义 run、FASTA、GO、settings,并对最后的 summary page 负责。
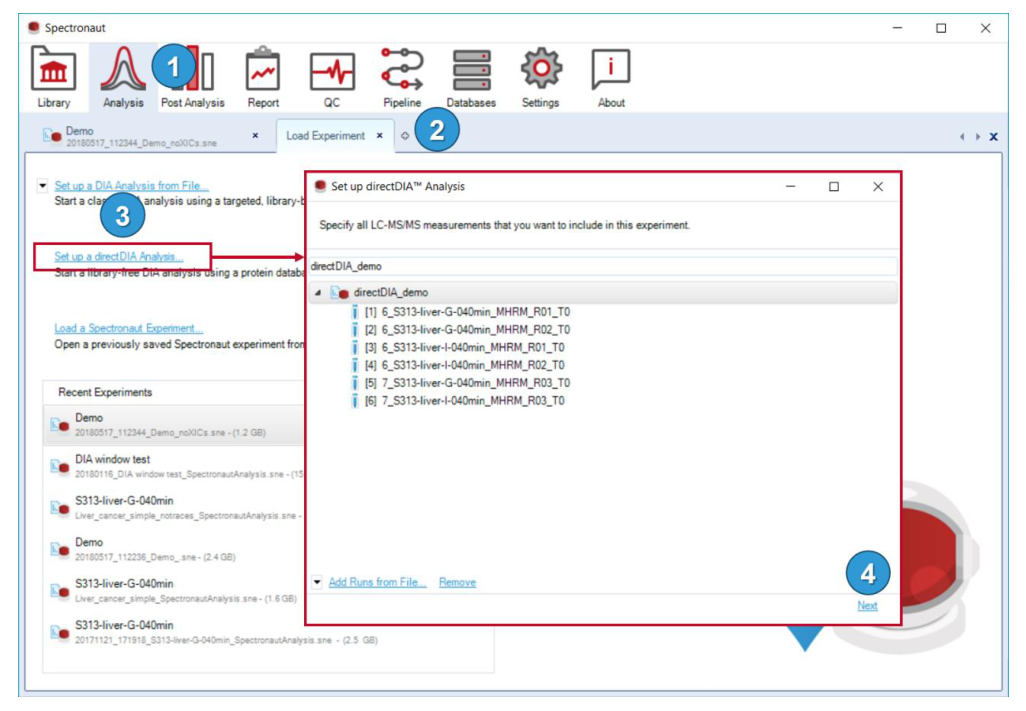
这一流程要求依次设置 run files、FASTA、GO annotation 和 analysis settings,最后在 summary page 再次确认。directDIA 只是绕过外部谱图库,不是绕过实验定义。
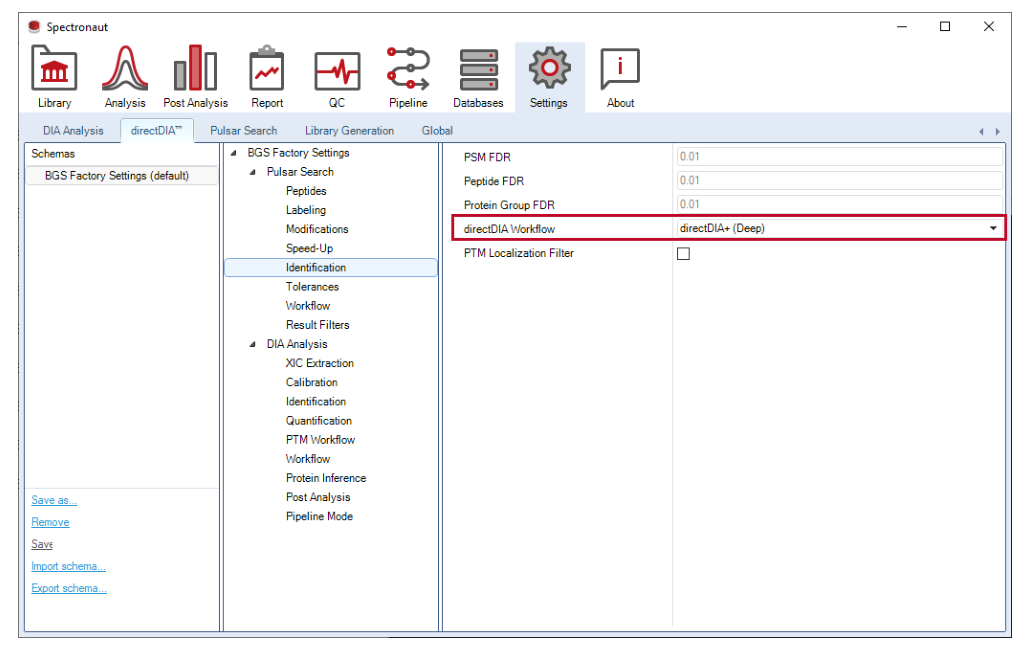
Fast 对应常规非 PTM search space,Deep 更强调覆盖。复杂 PTM 搜索空间下,二者相对收益会发生变化。
PTM & Method Evaluation
这两条路线直接改变搜索空间和结果解释方式,因此需要单独建立边界与使用顺序。
Spectronaut 20 引入 Kuiper 后,可以把 variable modifications 拆成 Known 与 Probe 两层,实现开放 PTM 探索。重点不是能搜更多修饰,而是 search space 变大后结果筛查和解释难度同步上升。
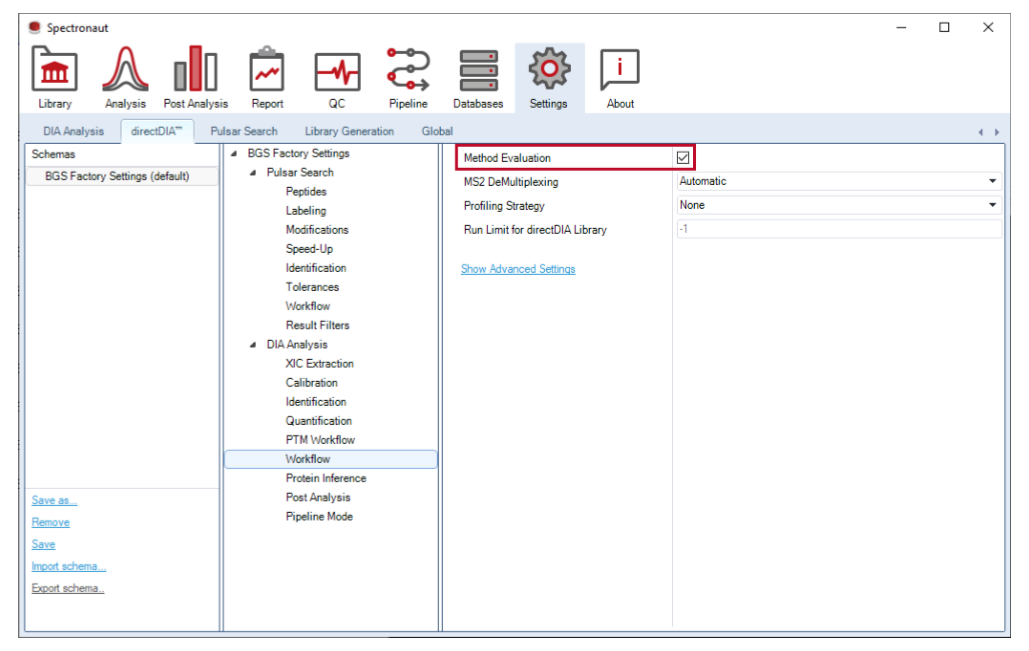
这一模式用于比较不同 DIA 方法,并会按 condition 分别搜索。它用于 acquisition benchmark,不进入正式定量主线。
Conditions
condition、replicate、fraction、reference condition 与 correction factor 共同定义统计结构,并直接进入后续差异解释。
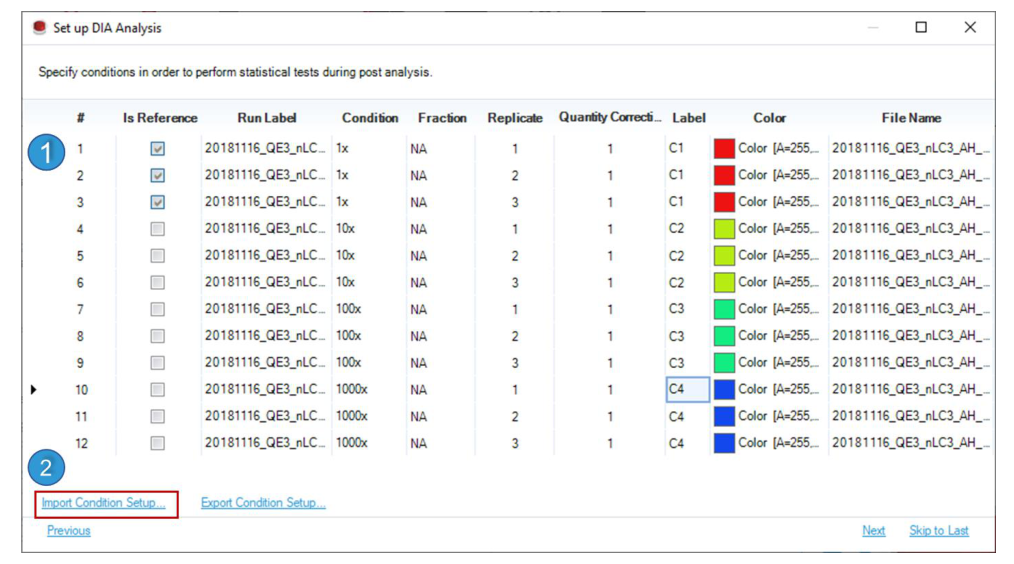
Label 列首先服务 plotting,决定后续 Post Analysis 的颜色标记;Is Reference 决定 differential abundance 的参考条件;quantity correction factor 则允许把最终 quantities 映射到初始样本体积等外部尺度。Advanced Workflows
Conditions 之后,分析结构开始从单通道 discovery 转入多通道定量、位点层定量和更复杂的对象层。这里最需要区分的,是 label-free、spike-in、plexDIA 与 PTM workflow 各自改变了哪一层证据结构。

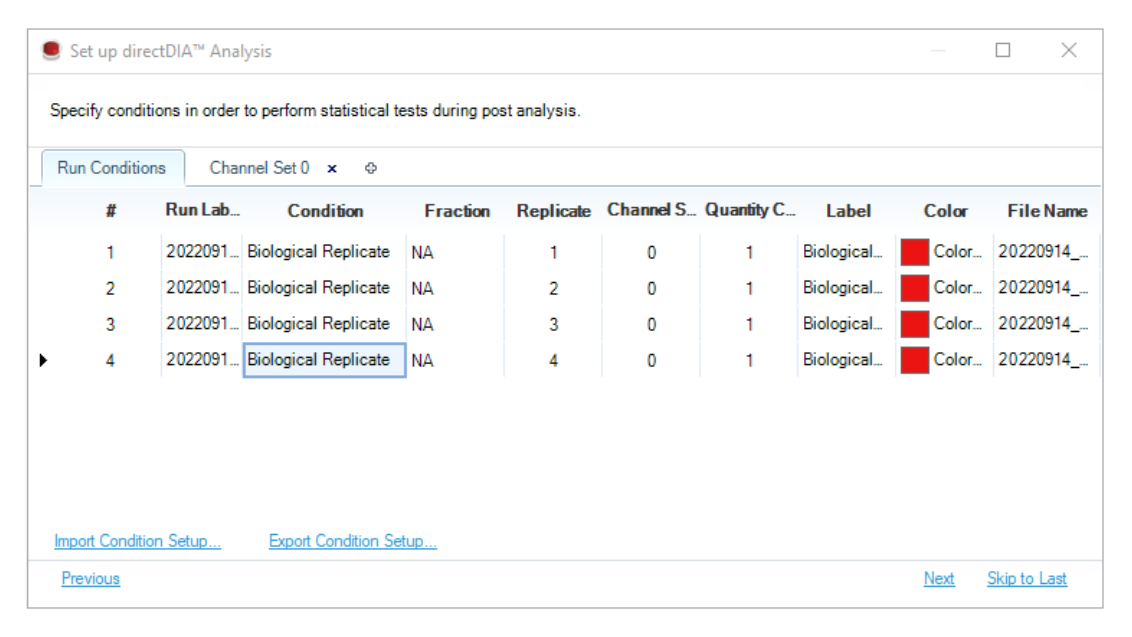

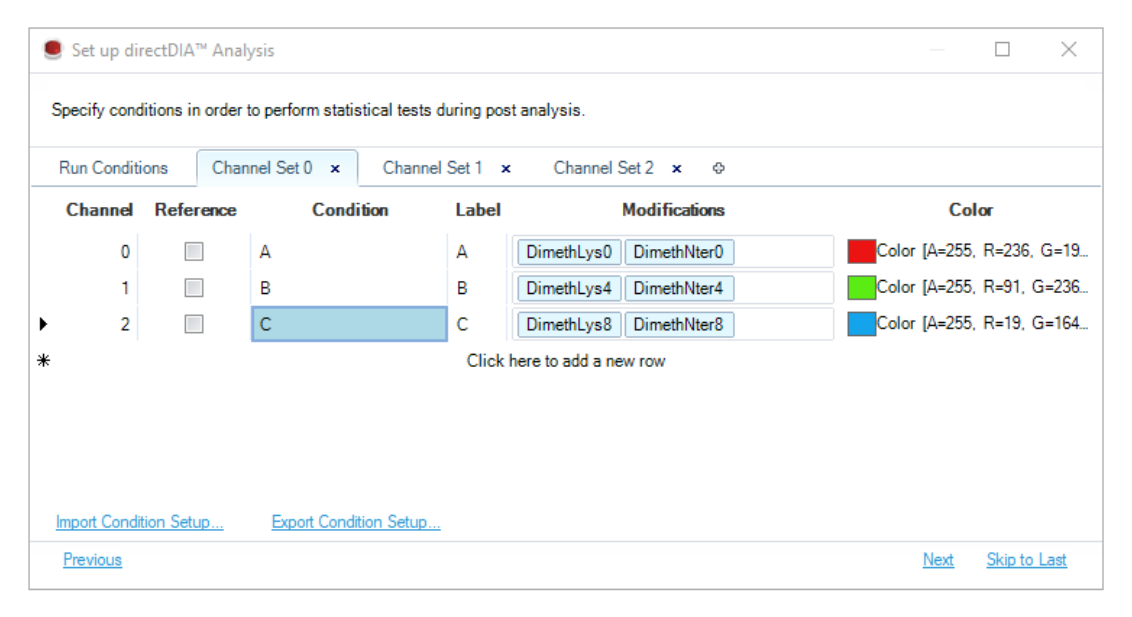
普通 labeled workflow 解决的是 reference ratio 或多通道读数进入同一分析结构;plexDIA 进一步把多通道 DIA 的共享峰边界、通道级定量和条件矩阵组织成正式 discovery 设计。两通道时,峰提取与打分会覆盖所有 channels;多通道时则必须用 channel sets 固定每个通道在实验中的真实含义。
Biognosys 在 Spectronaut 19 / 20 和 HUPO 2024 资料中反复强调,与 Yansheng Liu 团队合作推进的重点,是把 multiplex-DIA 用于 protein turnover 与 true abundance 的联合分析。要做到这一点,必须同时管理 channel sets、channel-specific FDR 和 heavy / light transition 选择。
PTM workflow 不只给位点列表。顺序是:先判断 modification occurrence,再给 localization confidence,再做 modification-site differential abundance;若启用 input normalization,则先按 reference sample 的 protein quantity 做归一;若启用 stoichiometry,则进一步计算 site stoichiometry。
| 结构层 | plexDIA 需要固定什么 | 如果没有固定会怎样 |
|---|---|---|
| Channel set | 每个 light / medium / heavy 通道在每个 replicate 中对应什么生物学条件 | 后续 ratio matrix、pairwise comparison 和 turnover time course 会失去解释基础 |
| 过滤层 | 整体保留还是通道独立保留,是否启用 channel-specific FDR | 弱通道被强通道掩盖,或者高覆盖结果被误当成稳定通道级证据 |
| Transition 层 | heavy / light 对应的 fragments 是否存在同位素 overlap 或干扰 | 定量看似完整,但真实 ratio 被 transition interference 拉偏 |
Review & Refinement
Grid View、Tree View、detached perspectives 和 rerun 能力,让 Analysis Perspective 不只是一次性执行器,而是一个可以回头修正实验定义和峰级判断的工作台。
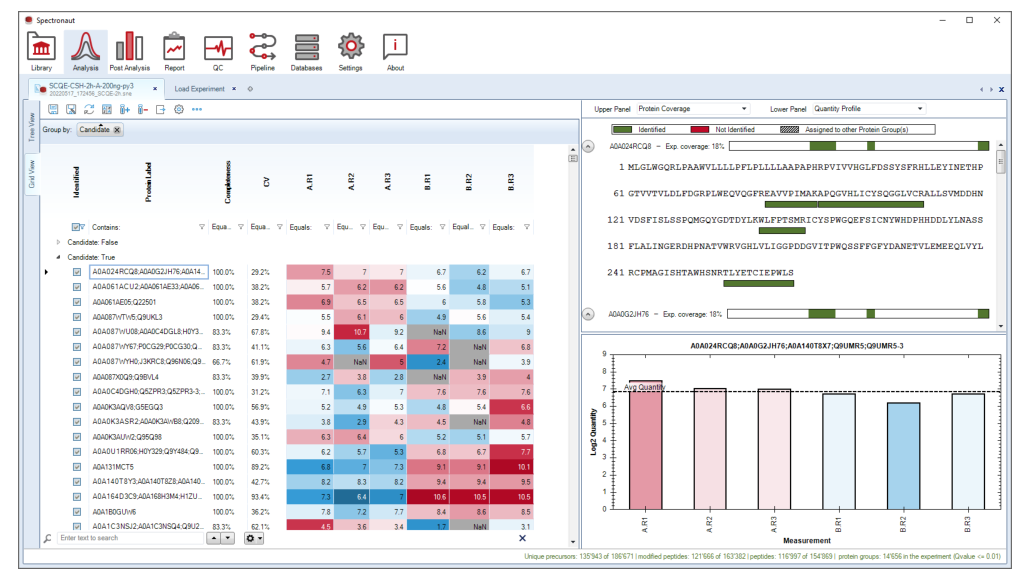
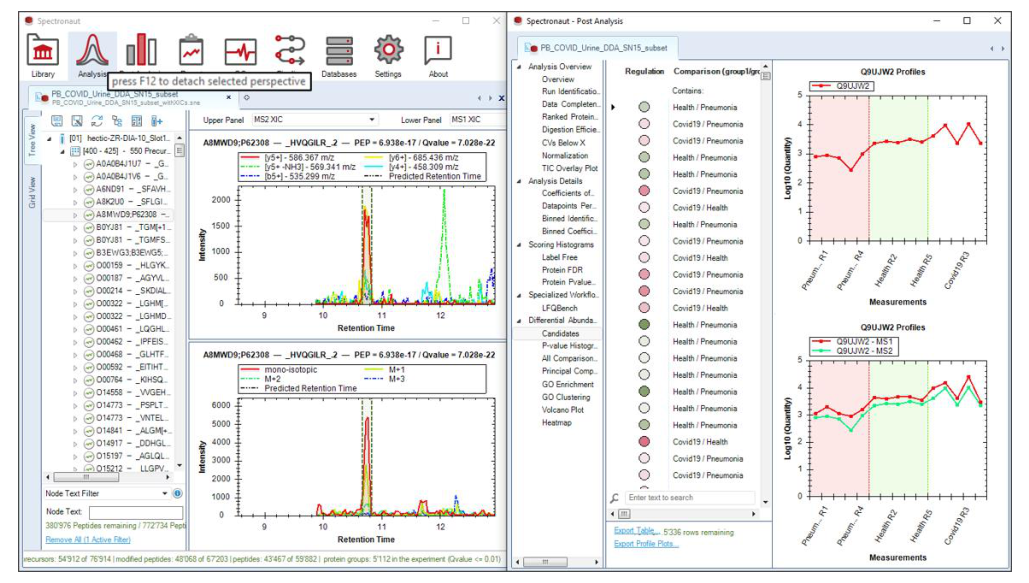
当统计上有意义但峰型存疑,或某批 run 明显异常时,不该只看表格,应回到 XIC、峰界、iRT 偏移和 interference 层重新确认。
如果问题来自条件设置、reference 选择或 settings schema,就应该 rerun,而不是在导出的 Excel 里事后补救。
.sne;不带 XIC 的文件更小,但重开时需要重新映射 runs。.sne 不带 XIC 则先 re-extract XICs,并可按 precursor window 对 tree 分组以提高 UI 响应。Refine Fragment Selection。在这里可以移除受干扰 fragments、加入覆盖关键 modification sites 的 ions,甚至引入原 DDA 中未检测但在 DIA 中清晰可见的 theoretical fragments。Commit Library Changes… 批量提交,随后触发所有相关 peptides 的 re-extraction。同时要保留 library version control,因此 refinement 不是覆写历史,而是带版本的可回退修改。directDIA JSON Settings
directDIA 项目进入命令行后,JSON settings 用于把 GUI 中的 workflow、识别阈值、定量策略、搜索空间和仪器容差明确写入文件。这样每一次批处理都有可复制、可审计、可回退的参数记录。
| JSON 区块 | 控制内容 | 分析含义 |
|---|---|---|
DIA_Analysis → Identification |
Precursor / Protein 层 PEP 与 q-value、single-hit protein rule、run-level protein scoring | 决定结果以什么可信度边界进入后分析和报告。 |
DIA_Analysis → Quantification |
proteotypicity filter、data filtering、cross-run normalization、imputation、IM peak picking、background noise removal | 决定跨样本定量矩阵如何过滤、归一化和填补。 |
Pulsar_Search → Peptides |
酶切规则、digest type、肽段长度、missed cleavages、N-terminal M 处理 | 决定 search space 的大小和半特异/非特异分析边界。 |
Pulsar_Search → Modifications |
固定修饰、可变修饰、最大可变修饰数、closed search 或 PTM probing search | 决定修饰空间和 PTM 探索压力。 |
Pulsar_Search → Identification |
PSM / peptide / protein group FDR、directdia_deep / directdia_fast / directdia、RT sampling reduction | 决定 directDIA 搜索强度、速度和识别阈值。 |
Tolerances / MS2 |
dynamic / relative / static 质量容差、MS1/MS2 correction factor、b/y/a/c/x/z ion types | 决定不同平台 raw data 的质量窗口和 fragment ion 证据层。 |
-j 导入的 JSON 会覆盖默认设置或 -s 指定的 .prop 设置。推荐流程是先用 GUI 导出基准 JSON,再在版本控制中只改项目必须改变的字段。
上线前必须检查 JSON 是否可解析、数值是否在允许范围、枚举值是否拼写一致、数组字段是否仍为数组,以及 DirectDIA_Workflow 是否明确。
相关专题
Reporting 与 QC 用于承接字段、schema、pipeline 与 XIC 证据层。
Report Schema、QC history、Pipeline 和 Appendix 8 / 9。