附录作为独立课程的理由
Spectronaut 的大量提取逻辑、图谱判读和参数影响都集中在附录里。跳过附录,就很容易只记操作路径,而缺少判断框架。
Spectronaut Appendix
本页把附录部分拆成一条独立的附录课程线,覆盖
DIA Analysis Settings、Pulsar Search Settings、
directDIA Settings、Library Generation Settings、
Analysis Perspective Plots、Experiment Tab Options 和
Post Analysis Plots。重点是把附录从“参数表”转成真正帮助建立判断的学习材料。
Orientation
最密集的设置和图谱说明被集中放进附录,是因为这些内容更适合在已经理解主流程之后再系统学习。附录因此承担进阶课程的角色。
Spectronaut 的大量提取逻辑、图谱判读和参数影响都集中在附录里。跳过附录,就很容易只记操作路径,而缺少判断框架。
附录分为两条主线:第一条是参数线,回答“软件如何决定提取、打分和定量”;第二条是图谱线,回答“结果为何可信以及哪里可能有问题”。
从参数附录到图谱附录,再到报表字段与 XIC 的学习衔接。
Appendix 1
Appendix 1 从 XIC 提取、离子淌度、质量误差容忍度、normalization、cross-run quantity、FDR 到 protein inference,完整说明软件如何提取峰并计入最终定量。
Dynamic、Static、Full 三种提取窗口不是“哪个更高级”,而是对可变性和搜索范围的不同取舍。默认 dynamic 的核心,是让软件根据 calibration 自适应窗口宽度。窗口越宽,不一定越安全,反而可能引入更多干扰。
Dynamic tolerance 表示由 Spectronaut 根据校准结果自动决定;Relative 和 Static 则意味着人为更强的约束。mass tolerance 的意义不是“调大就更容易识别”,而是在灵敏度与特异性之间找到平衡。
因为 normalization、cross-run quantity 和 protein inference 并不只是分析后处理,而是从一开始就影响结果矩阵如何被构建。它们决定了 data completeness、CV、fold change 和 differential abundance 的上限。
在这一组设置里,identification threshold、quantification threshold、precursor 层 q-value、protein inference 与 single-hit protein 处理不是分散的按钮。它们共同决定哪些信号可以进入对象集合、哪些对象可以进入定量矩阵、以及蛋白层结果最终能否稳定交付。对 discovery DIA 来说,precursor 层控制得越细,越需要在蛋白层同步说明 inference 与汇总边界。
| 设置点 | 直接改变什么 | 放宽或改变后最容易出现的后果 |
|---|---|---|
| Identification threshold | 哪些 precursor 可以进入已识别对象集合 | target / decoy 分离尚未站稳时,后续所有对象层都被放大 |
| Quantification threshold | 哪些 precursor 可进入定量矩阵 | 矩阵 completeness 看似更高,但低质量读数会抬高 CV 与噪声 |
| Protein inference | 哪些 peptide / precursor 被汇总进 protein group | 同名蛋白组在不同项目间失去可比性,蛋白层名录随规则变化 |
| Single-hit protein | 单条证据支持的蛋白是否保留 | 蛋白层数量上升,但经验错误比例和交付边界同步变宽 |
precursor 层一旦允许更多对象进入,蛋白层的风险不会自动保持不变。protein inference 会重新组织 shared peptides 与支持证据,single-hit protein 则会决定单条证据是否直接进入 protein group。也就是说,蛋白层结果的大小与稳定性不是报表阶段才决定,而是在 Appendix 1 这一层已经被部分写入。
如果数据库同时引入 canonical 与 non-canonical FASTA,或 workflow 进入 peptidomics、开放修饰和更宽的 search space,蛋白层压力会进一步上升。此时最稳的做法不是只盯 precursor q-value,而是同步固定 database 版本、group 结构、protein inference 规则和 single-hit protein 策略。
Appendix 2-4
这三个附录经常被分开看,但从学习角度更好的方法是把它们视为一组:Pulsar 决定搜索空间和建库逻辑,directDIA 决定 library-free 的分析逻辑,Library Generation Settings 则决定 project-specific library 如何被构建和复用。
这部分需要重点强调酶切、修饰、search mode 和过滤策略如何共同决定 Search Archive 的内容。这里不应被孤立成“建库参数”,而应视为后续所有 library 质量的上游来源。
directDIA 的核心不是“没有 library”,而是“由 raw + FASTA 自己生成项目知识库”。因此它的 settings 应和 Pulsar search settings 对照着讲,帮助新人建立“两步式”分析理解。
这一附录决定 library generation 过程中的 protein inference、library-wide FDR、QC kit 派生能力和 GO 相关信息。它可与 Library Perspective 一起复盘。
Pulsar、directDIA 与 Library Generation 共同决定 search space 的大小和结构。canonical FASTA、non-canonical FASTA、开放修饰、unspecific 搜索或 PTM probing 一旦把候选空间推得更开,错误发现控制就不能只停在统一阈值,而要进一步进入 precursor 层与分组层控制。SP20 的 global precursor FDR 与 precursor FDR per group,正是在这一段开始真正产生价值。
| 数据库 / workflow 结构 | search space 会怎么变化 | 最该同步固定的规则 |
|---|---|---|
| Canonical FASTA + library-based DIA | 候选空间最稳定,group 结构较容易保持一致 | 固定 precursor threshold、protein inference 与常规 single-hit 策略 |
| Canonical + non-canonical FASTA | 前体候选和蛋白分组明显增加,shared evidence 更复杂 | 启用 per-group precursor FDR,并写清 group 结构与蛋白层汇总方式 |
| directDIA + open / unspecific / PTM probing | 项目内自建知识库,候选空间更开放 | 同时固定 database 版本、search mode、protein inference 与 single-hit protein |
当项目把数据库从 canonical 扩展到 canonical + non-canonical 时,软件层面至少有四个地方必须同步改写:precursor 层错误发现控制、group 结构、protein inference 汇总方式,以及 single-hit protein 的保留策略。只要其中任意一项继续沿用旧项目设置,蛋白层名录和经验错误比例就可能失真。
SP20 的 global precursor FDR 与 precursor FDR per group 需要和 protein-level 规则一起阅读。前者解决更开放搜索空间下 precursor 层的稳健控制,后者决定这些 precursor 如何进入最终的蛋白层交付对象。
Appendix 5
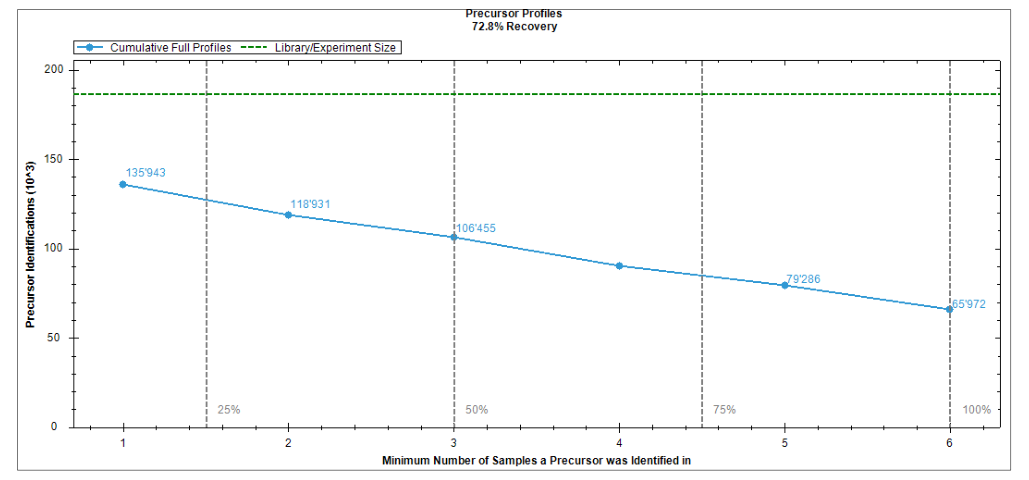
Appendix 5 是最像“读图教科书”的部分。从 iRT calibration 到 PDM,再到 XIC alignment 和 protein coverage,这些图共同构成了 Analysis review 的证据体系。
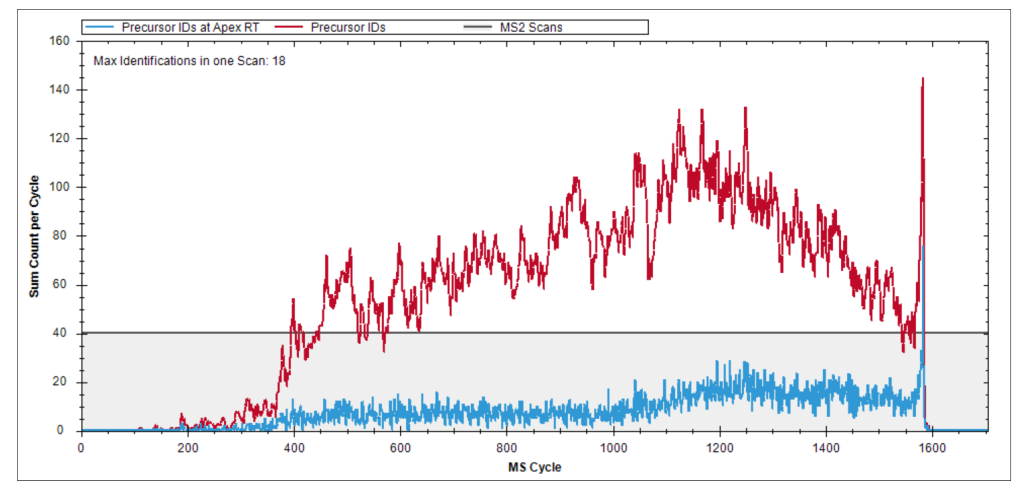
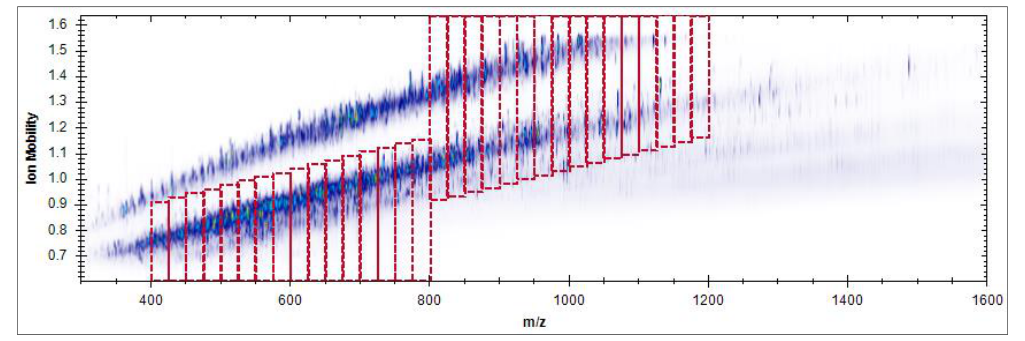
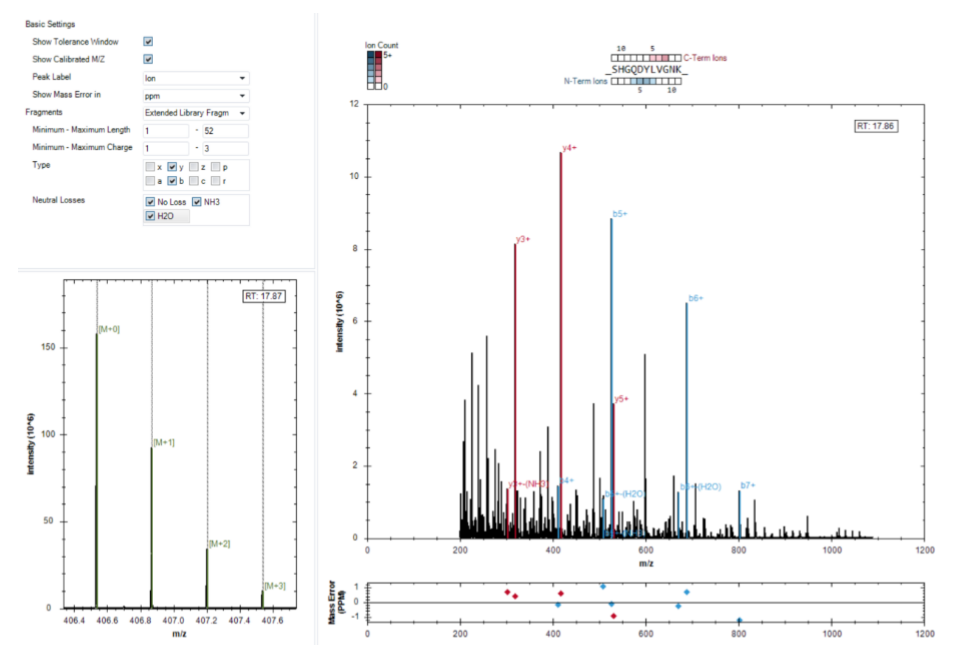
不需要一开始就把二十多张图全部看完。更稳的节奏是先理解 iRT calibration、identifications per cycle、PDM 和 XIC alignment 这四类,它们最容易帮助建立“好图”和“坏图”的直觉。
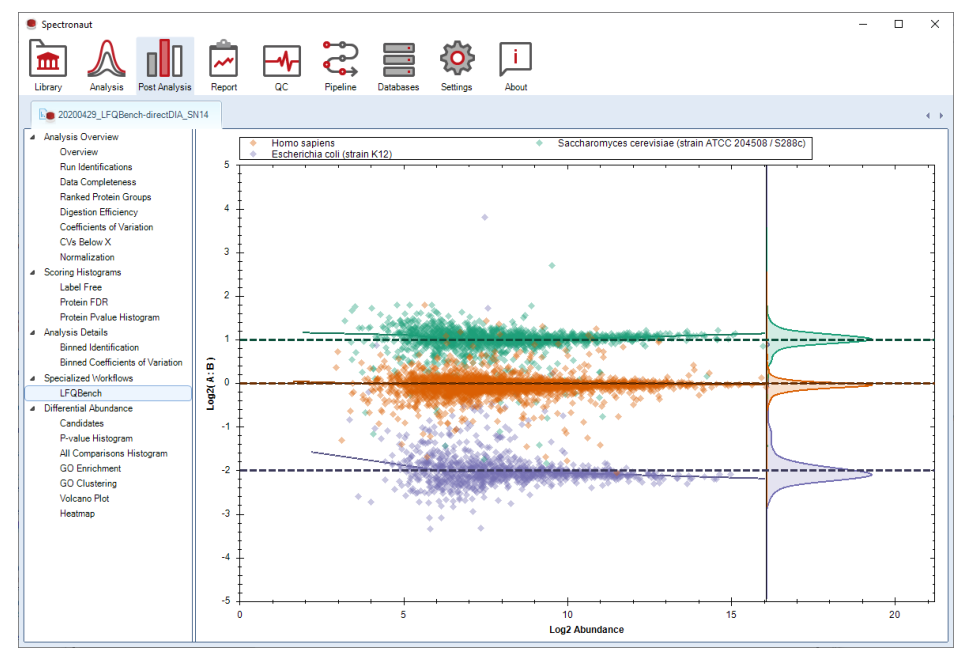
Ion mobility overview、DIA acquisition method overview、precursor vs protein quantities 这些图,已经提供 acquisition method 优化和验证的窗口,因此它们不只是分析后复核工具,也能进入方法开发学习。
Appendix 6
这部分容易被低估,但它非常适合作为上机实操课。accept、reject、hide、exclude、pending changes 这些操作会分别改变对象状态、显示结构和后续结果。
实验 tab 里的手动操作不是“随便改一改图”,它们会改变 review 状态、可见性乃至定量输入。这里应始终结合 `IsUserPeak`、`IsVerified` 这类字段一起理解,避免 GUI 操作和报表输出脱节。
当样本异常、峰界可疑、某一批次整体偏移时,Experiment Tab 是最快的排查入口。因此它不只是“操作说明”,也是交付前最后一层人工把关机制。
Save / Save as、Group by、Order Runs By 这类更偏组织和浏览;而 Process Pending Changes、Refresh Post Analysis、Commit Library Changes 则会真正影响分析结果或其解释层。Appendix 7
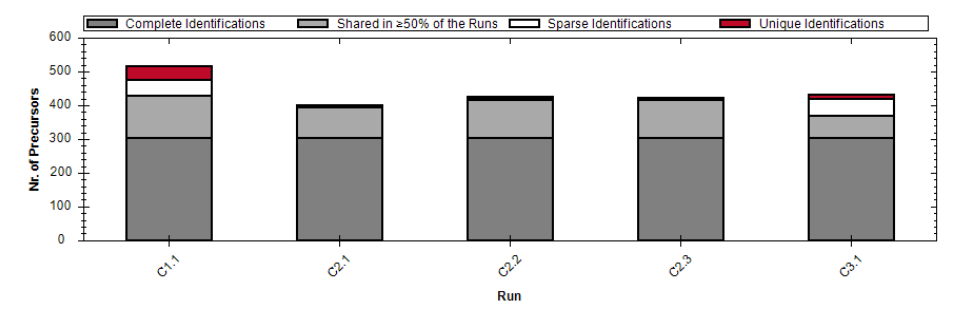
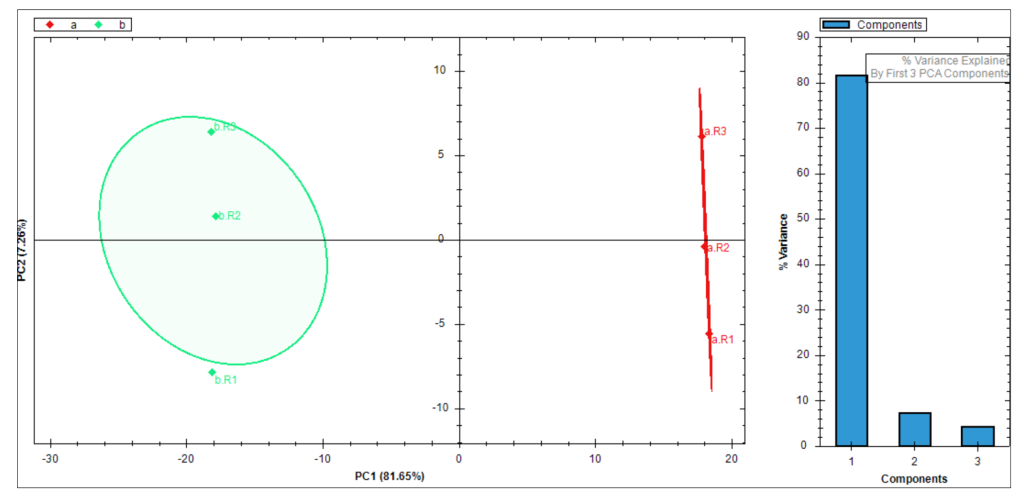
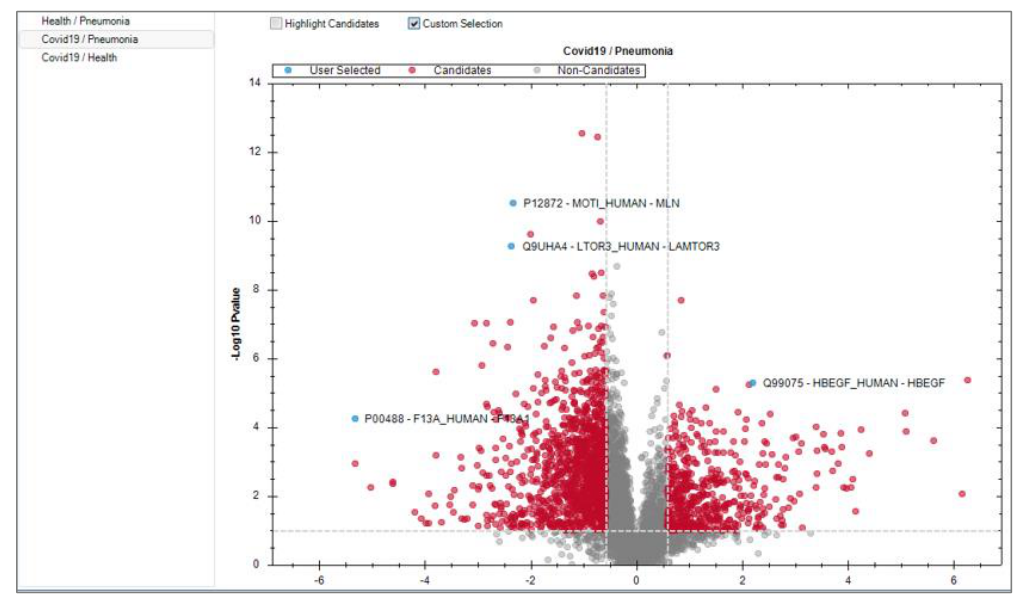
从 run identifications、data completeness、CVs below X 到 LFQBench、PCA、volcano、heatmap,Appendix 7 其实已经构成了一整套“结果复核课”。
Bridge
更稳的顺序不是一直停在图和参数,而是在理解了参数和图谱之后,进入字段词典和 XIC 数据库接口。这样“如何运行”“如何导出”“如何复核”就能完整接起来。
继续学习 Appendix 8 的对象层级字段字典,以及 Appendix 9 的 XIC Export DB。
把 precursor 层控制、protein inference、single-hit proteins 与经验错误比例连成一条交付链。
如果需要更快查看附录核心 figure,可从软件页的 Appendix 图谱入口直接进入。
Next Steps
可按 Analysis 主流程、附录图谱、Reporting / XIC、字段练习与排错四层连续查看,使参数、图谱、字段和交付结构保持一致。
把附录参数和 workflow 重新对应起来。
继续学习 Appendix 8 / 9 与报表对象层级。
继续查看所有章节、附录索引和更完整的细译块。