结果解析的作用
结果解析先处理矩阵质量、差异结构和候选筛选,再进入具体报告和结果交付。
结果解析
结果解析模块覆盖
Analysis Overview、Scoring Histograms、Candidates、
PCA、GO Enrichment / Clustering、Heatmap、
Volcano 和 PTM analysis,对应从结果矩阵到机制解释的完整流程。
总述
结果解析部分覆盖 identification、quantification、differential abundance、hierarchical clustering、PCA、GO enrichment 与 PTM 位点结果,对应 discovery 项目从结果矩阵走向生物学解释的正式入口。
结果解析先处理矩阵质量、差异结构和候选筛选,再进入具体报告和结果交付。
结果解析负责解释当前发生了什么,报表模块负责组织后续交付。如果前一层没有建立清楚,后一层就容易变成导出很多列却无法解释。
从矩阵质量、候选筛选到 GO / PTM 解释的主线结构。
总览
Overview 会展示 proteins / peptides identified and quantified、missed cleavages、library recovery,以及 identifications、data completeness、CV、normalization、detected modifications 等多种图。这意味着在看任何候选之前,我们就已经有足够的信息去判断这次实验是否稳定。
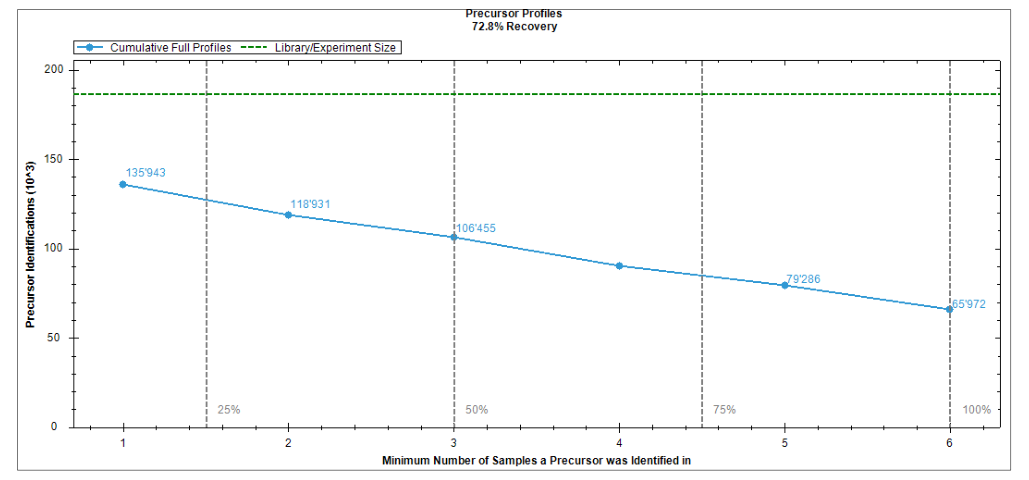
最好的顺序是先看 identifications 与 quantified objects,再看 completeness、CV 和 normalization,最后再回头理解 detected modifications 和 peptide motif plots。这样你会先建立“矩阵质量观”,而不是直接跳到某个显著蛋白。
它不是一张内部算法图,而是直接显示 target / decoy 分布、Cscore、q-value 和 precursor-level sensitivity 的关系。这一层会直接影响“同一个 fold change,是否具备相同可信度”的判断。
候选与 PCA
Candidates table 默认带有 q-value 和 fold change 过滤,而 PCA 用于观察样本是否按蛋白组轮廓分群。这两者一张偏“筛选与注释”,一张偏“全局结构”,应配合阅读。
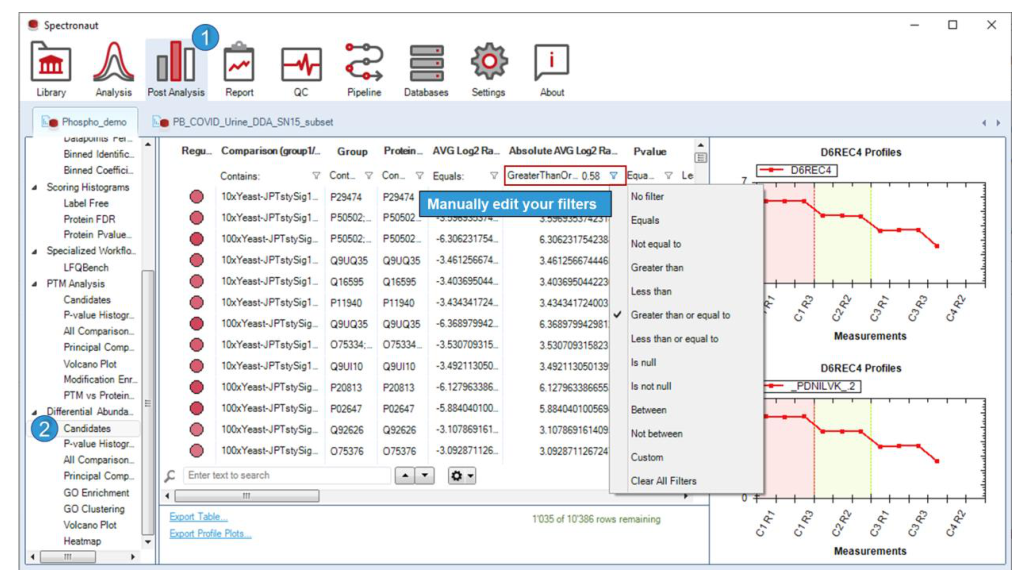
这里明确说明 q-value 和 log2 ratio 过滤会联动到 volcano plot;Column Chooser 允许你把 p-value 等字段加回来。也就是说,这张表不是静态输出,而是可交互的候选定义器。
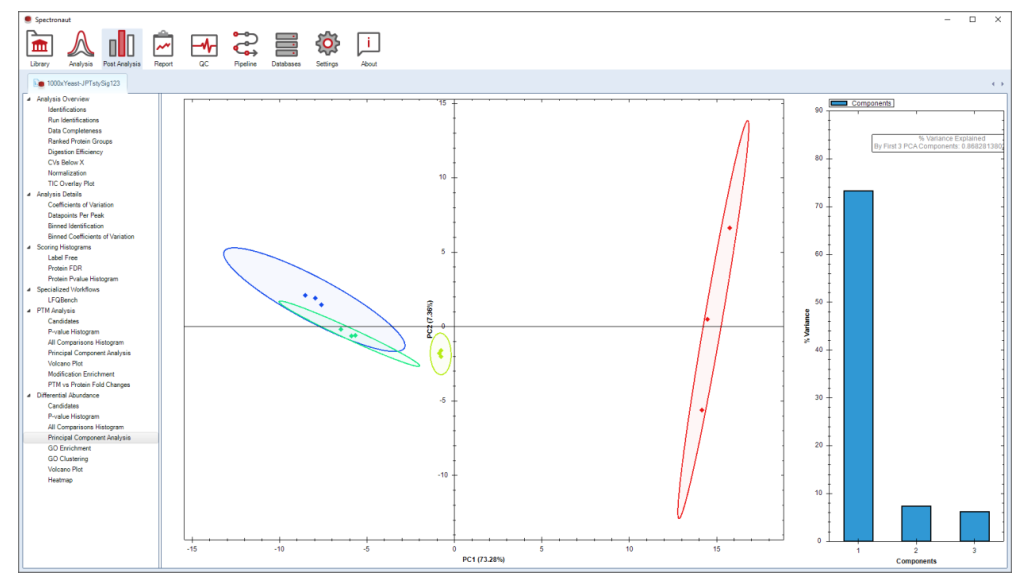
PCA 不是为了找“显著蛋白”,而是为了确认样本是否按预期分群、是否存在批次异常、前几主成分解释了多少方差。它最直接的作用,是在深入解释之前先看清全局结构。
GO 富集
第 71-74 页的重点不是“会跑 GO”,而是知道 background proteome、候选集、over / under representation、multiple testing correction 和 term similarity cutoff 各自意味着什么。否则 GO 图很容易变成“看起来很热闹、解释却很松散”的部分。
必须先理解 background proteome 是全实验中识别到的蛋白,而不是整个 FASTA;再理解 Bonferroni 与 Benjamini-Hochberg 的差异;最后再看 enrichment table 怎么过滤、怎么重算。这样你才不会把 GO 看成一个黑箱按钮。
当 enrichment 结果很多时,就需要 clustering 来降低冗余。REVIGO 风格的语义相似度聚类,正是把“很多显著 GO term”压缩成“几个更可解释主题”的关键步骤。
图形
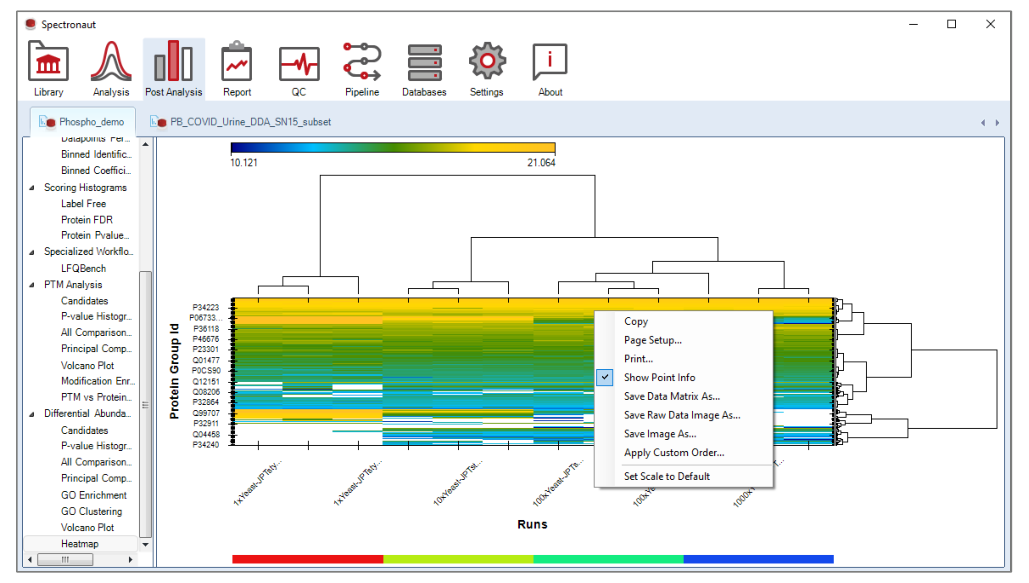
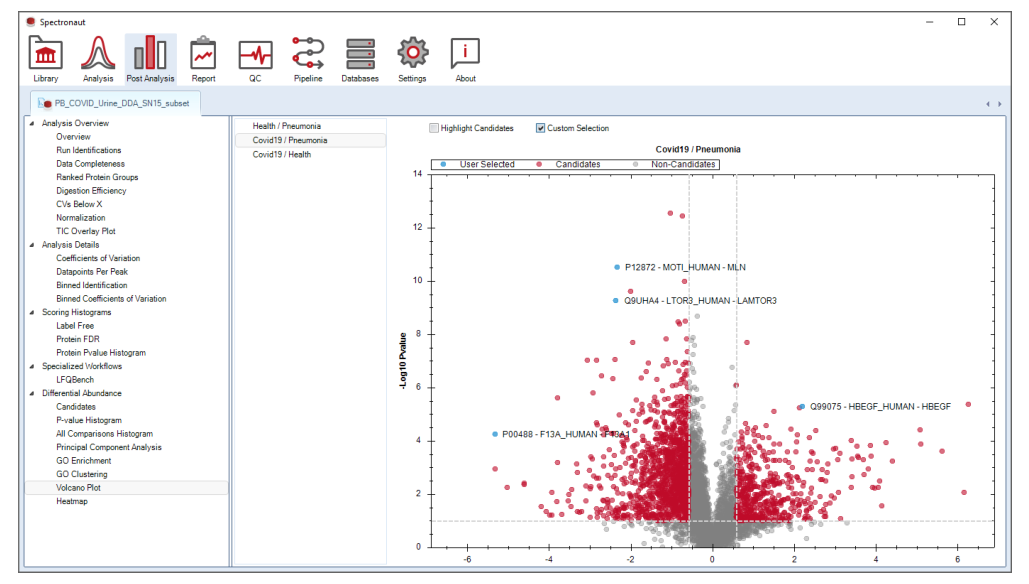
Heatmap 和 volcano 是最重要的 post-analysis plots。Heatmap 强调行列聚类与 matrix export,Volcano 强调候选集与阈值联动。这意味着它们都应该被理解成“和筛选逻辑紧密耦合的图”,而不是独立的可视化终点。
PTM
当启用 PTM workflow settings 时,Spectronaut 会在 modification site level 做差异分析,并提供 Modification Enrichment 与 PTM vs Protein Fold Changes。关键是区分“蛋白变了”与“位点调控变了”。
这里说明 modification site object 的唯一标识在 Group 列里,这一点可直接用来解释“位点层对象”和“蛋白层对象”不是一回事。
Modification Enrichment 对应富集方向和位点类型分布,PTM vs Protein Fold Changes 则直接回答“位点变化能否脱离蛋白总量变化独立存在”。这是修饰组里非常核心的视角。
Group 列给出 modification site object 的识别键,Modification Enrichment 则显示各实验 run 中携带该 modification 的 precursor 百分比,PTM vs Protein Fold Changes 则识别独立于蛋白总量变化的位点调控。相关专题
报表与 QC 承接字段输出和质量解释,附录图谱用于复核后分析图形和参数边界。