核心结构
这里围绕四个结构展开:setup wizard 里 raw file、panel、calibration curves 与 analysis schema 的关系;absolute quantification 对 workflow type、reference channel、LLOQ / ULOQ 规则的依赖;树状 review、grouping 和 filtering 的方法开发作用;以及 refinement 回写 panel 的正式路径。
SpectroDive Analysis
这里覆盖 targeted analysis、condition annotation、absolute quantification、
review / grouping / filtering、panel refinement 和 iRT refinement 的主分析链,对应峰证据、transition 保留、定量边界与 panel 回写的完整关系。
Orientation
Prepare Perspective 决定 panel、RT calibration 和导出方法;Analysis Perspective 决定这些设计是否在真实数据里站得住。这里不仅承担 targeted data analysis,也承担 scheduling analysis、panel iRT refinement analysis,并且支持加载保存的 SPE 实验继续复核。
这里围绕四个结构展开:setup wizard 里 raw file、panel、calibration curves 与 analysis schema 的关系;absolute quantification 对 workflow type、reference channel、LLOQ / ULOQ 规则的依赖;树状 review、grouping 和 filtering 的方法开发作用;以及 refinement 回写 panel 的正式路径。
SpectroDive Analysis 直接承接 Spectronaut discovery 之后的 targeted panel。候选一旦进入 targeted panel,这里就负责确认其是否可验证、是否可定量、是否可纳入 SOP。
从 experiment setup 进入 review、absolute quantification、panel refinement 和结果交付的工作链。
Analysis Perspective 包含四项正式任务。第一,建立 targeted data analysis,把真实 run、panel 与 analysis schema 结合起来完成定量和识别。第二,建立 scheduling analysis,服务 method export 前后的调度验证。第三,建立 panel iRT refinement analysis,把实际采集结果回写到 panel。第四,加载保存的 SpectroDive experiment 继续复核。
这一章的核心是分清对象类型。raw file、panel、analysis schema、calibration curves file、condition setup 属于前置输入;review judgement、refinement changes、saved experiment 属于分析阶段沉淀出来的文件。把这两类对象分开之后,绝对定量和 refinement 的逻辑才会稳定。
Setup
Experiment Setup 中的每个输入都会影响后面结果的可解释性。Raw file 决定信号来源;panel 决定提取对象;calibration curve files 决定是否做绝对定量;analysis schema 决定提取、打分和 FDR 的底层规则;condition annotation 决定后续 differential abundance 和 condition-wise metrics 的统计骨架。
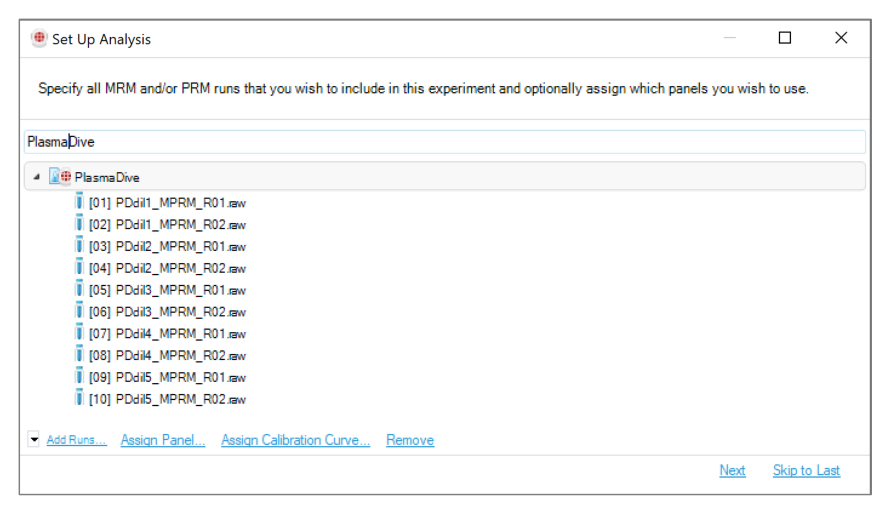
单次方法开发或练习场景,用文件入口更直观;成批 PRM / MRM 运行,用文件夹入口更接近真实项目。这两种入口分别对应单次 run 与批量 run 的不同组织方式。
BGS Factory Settings 可覆盖大多数实验,但并不等于所有项目都应长期停留在默认值。schema 本身就是软件级 SOP,默认值与覆盖规则需要一起管理。
第 41 页特别提醒 conditions editor 对大小写和空格敏感,这表面上像输入细节,实质上说明 condition 注释不是装饰字段,而是会直接进入 differential abundance 的统计对象层。
系统会先检测 iRT kit、据此校准分析参数,并利用它提升处理速度、specificity 与 automated QC。因此 iRT kit 是 scheduled analysis 和自动 QC 的基础设施。
这段向导可以按 6 个连续问题来学。第一,你准备分析哪些 run,也就是从 file 还是 folder 导入 raw files。第二,你准备用哪个 panel 去提取它们,这决定了软件会去哪些 precursor 和 transition 上找信号。第三,你是否要启用 calibration curves,如果是,就必须同时指定一个或多个 .ccs 文件。第四,你这次分析打算采用哪套 analysis schema,也就是 Identification、Quantification 和 Post Analysis 的底层规则。第五,你的实验条件和重复设计是什么,这一步由 Condition Setup 承担。第六,你是否准备好让软件开始根据 iRT kit 做自动校准,点击 Finish 后这一流程会立即开始。
Condition Setup 的内容比很多人想象中更细。它不仅包含 condition、biological replicate 和 fraction,还包含 Run Label、Is Reference 与 quantity correction factor。前两个字段主要进入 plotting 和 reference-based normalization,后一个字段则会直接乘到最终数量上。也就是说,Condition Setup 并不只是为了做一张颜色漂亮的后分析图,它其实是整个统计设计的输入层。
Condition annotation 提供 3 条导入路径:使用 file name parsing schema 自动从文件名解析、直接在表里手工编辑、先导出当前 setup 再修改为文本文件重新导入。自动解析对应标准化批处理,手工编辑对应小规模试验,文本导入对应固定模板项目。conditions editor 对空格和大小写敏感,因此条件命名会直接影响统计分组。
点击 Finish 后,系统会先自动检测 iRT kit,并按每个 run 的数据特征校准分析参数。这一步直接决定处理速度、specificity 和 automated QC,因此底部 progress bar 完成初始校准之后,才进入稳定浏览状态。
Calibration
这一部分把标准品序列、calibrant runs、workflow type、reference channel、dilution factor、LLOQ、ULOQ、LOD 和线性范围连成一条闭环。这里的核心不是单独导入 .ccs 文件,而是理解它所承载的定量模型。


校准曲线的价值,在于通过已知浓度标准样本和未知样本之间的关系,把“相对信号”变成“带单位的浓度估计”。但只有测得信号落在 LLOQ 到 ULOQ 的线性范围内,这个绝对定量才是精确且可报告的。
当 light endogenous peptides 保持恒定浓度,而 heavy 标准作稀释系列时,应选 inverted spike-in workflow,并让 light peptide 作为 reference channel 参与识别。也就是说,workflow type 不是 nomenclature,而是定量模型本身。
当复杂基质里的目标肽段长期徘徊在 LOD 附近时,单靠 direct PRM 往往无法稳定进入可报告的线性区间。SISCAPA 通过抗肽抗体免疫富集降低背景噪声,再由 SpectroDive 承接 calibration curves、LLOQ / ULOQ 和报告字段。
LLOQ 是可接受准确度和精密度的最低可定量点,不等于刚刚看得见。LOD 是检测下限,表示可以检出,但并不等于可以稳定定量。ULOQ 是仍保持线性响应的最高可定量点。AbsoluteAmountRangeLimitted 这种字段的意义,在于提醒你“有数值不代表在可报告区间内”。dilution factor 和样本准备历史必须进入计算链,否则绝对量会失真。.ccs 不是普通附件,而是 calibration curve set 的正式文件,应该像 panel 一样被版本化管理。这一套 calibration curves workflow 必须按步骤理解,而不是只记住“导入一个 .ccs 文件”。第 1 步是先搭建一轮专门用于生成 calibration curves 的 targeted analysis:选择 calibrant runs,并指定本次分析对应的 protein panel。第 2 步是在 Workflow 节点定义 experiment type,例如当 light endogenous peptide 保持恒定浓度、heavy 标准做稀释系列时,应采用 inverted spike-in,并让 light peptide 成为 identification 的 reference channel。第 3 步是在 calibration curve 节点勾选 Process as Calibration Curve,并检查 Concentration variation on peptide type 的设置是否与实验设计一致。第 4 步是录入每条 peptide 的标准量信息;如果使用 Biognosys panel,一部分起始量可自动带入。第 5 步是定义 condition setup,尤其是 blanks、未稀释样本和各个稀释倍数的 dilution factor。第 6 步是在 Analysis Perspective 中复核生成结果,并把 calibration curve set 保存成 .ccs 文件。第 7 步才是启动真正的 quantitative experiment:加载待定量 run、指定相同 panel 和刚保存的 .ccs 文件。第 8 步是在 quantitative analysis 里保留 workflow type 的设置,但跳过 calibration curve generation 节点,因为这一步已经在前面的 calibrant analysis 中完成了。
第 6 步生成的是定量模型,第 7 步使用的是定量模型。前者输出 .ccs 文件,后者输出未知样本的绝对定量结果。把这两步分开,同一套 calibration curves 就能被多个实验重复使用,several calibration curves 也能为不同 precursor 选择最优曲线。
校准曲线 revision 这一页可直接按图阅读。绿色菱形表示该 calibrant run 中被高置信识别到的肽;x 轴是基于起始量和 dilution factor 计算得到的 nominal concentration;浅蓝色方框是通过校准曲线反推得到的实际定量值;低于 LOD 的点会以橙色框表示,提示它已经落到噪声底;红色虚线表示 blanks 的平均强度,对应背景噪声与信号分离程度。
如果在 Condition Setup 中定义了 replicates,图上还会出现统计离群值的三角标记,垂直误差线则反映每个 calibrant level 的强度 CV。读图时应同时检查三点:该 precursor 在 LLOQ 和 ULOQ 之间是否有足够多的点支撑线性区间;blanks 是否足够低;quantified value 是否稳定落在可报告范围内。
Review
SpectroDive 的 review 不只是“看一个峰”。它要求在 experiment tree 里按结果对象层级组织视图,并通过 Accept / Reject / Exclude / Hide / Delete 等操作对结果做明确判断。对 PRM / MRM 来说,这种审阅本身就是方法开发的一部分。


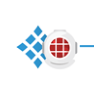
当项目从单一 run 进入多样本、多 panel、多 workflow 时,review 的难度会急剧上升。按 run、target、workflow 或 panel 分组,能够把复杂结果重新切回可人工判断的块。
接受、拒绝、排除、隐藏、删除这些动作不能混着用。有些动作只影响当前显示,有些动作会影响 quantification,有些动作会被提交进 refinement。区分这些差异,才能避免 panel 和结果同时失控。
默认 tree hierarchy 为 Run → Panel → Elution group → Precursor → Transition。Accept 和 Reject 针对 identification judgement:一个 elution group 被手工接受后,会被当作已识别对象;被手工拒绝后,会被当作未识别。Exclude 更偏结果与计算层:排除 elution group 或 transition group 后,它不会出现在 report 中;排除 transition 后,它既不会参与 quantification,也不会参与 scoring。Hide 只影响当前图形显示,不改变定量和报告;Delete 会把对象从 panel 中彻底移除。
排除或删除 fragment 后,父级 elution group 不会立刻重打分。要让修改真正回到 score 和 q-value 上,必须在 experiment tab 中点击 Process Pending Changes。
Tree filtering 与 tree grouping 是另一层组织能力。Filtering 只影响 Analysis Perspective 中显示哪些对象,不会自动改变 Post Analysis;Grouping 用来把复杂结果按 protein 或 panel 等方式重新组织。review 前应先确认 filters 是否启用,再确认当前分组方式。
Refinement
Refinement 不是分析结束后的附加步骤,而是 panel 迭代的正式机制。任何 targeted workflow 进入标准服务后,都要依赖这一章完成方法更新。
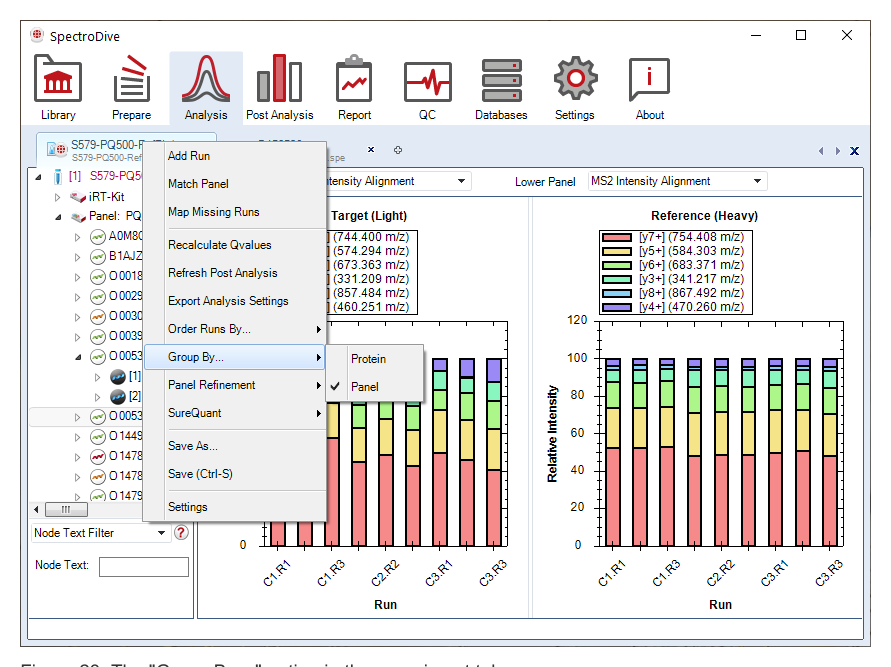
它允许在真实数据上判断哪些 transitions 更适于定量、哪些更适于身份确认、哪些应被淘汰。针对 PRM,还可以从理论 fragment 中继续筛选,因此 SpectroDive 不只是消费 panel,而是在生成更优的 panel。
任何 refinement 都必须通过 commit changes 真正回写到 Prepare Perspective。对于从 SPE 文件加载的 SureQuant / HybridDIA 实验,还需要先 re-extract raw data。
iRT Refinement run type,而不是拿普通 analysis 方法硬改。无论是 fragment selection refinement 还是 iRT refinement,本质上都属于 panel refinement,而 refined panel 会回写到 Prepare Perspective。也正因为如此,Prepare 中才提供 Reset to Factory Default 作为回退入口。
Fragment Selection Refinement 的重点在于“用真实采集结果筛掉不理想的 transitions”。PRM 不仅可以从 library 中已有 fragment 里筛选,也可以把 theoretical fragments 纳入候选,这意味着 PRM 方法优化并不只是在现有 panel 中做被动取舍。执行变更时,必须通过 experiment tab 的 Refine Assay Transitions 提交;如果是从 SPE 加载的 SureQuant 或 HybridDIA 实验,还要先执行 SureQuant → Re-extract Raw Data,否则这些变更不会体现在后续 refinement 中。
iRT Refinement 是 panel 迁移和方法早期开发的关键工具。先在 Prepare Perspective 导出专门的 Run Type = iRT Refinement 方法;这类方法会自动采用更宽的窗口,并默认每条 peptide 只保留两个 transitions。采集完这些 runs 之后,再用 Set up Panel iRT Refinement Analysis 建立分析,并在 experiment tab 中执行 Refine iRT。只有手工接受的 peptides 会进入 iRT refinement,完成 refinement 后仍需 commit panel changes。
SpectroDive 支持的 quantitative workflows 包括 label-free、labeled、spike-in 和 inverted spike-in。四者在 peak detection、scoring 与 post analysis quantification 的参考通道上并不相同,因此 workflow type 会继续影响 calibration curves、report 字段和结果解释。
SpectroDive 不会自动保存分析结果,因此需要主动执行 Save As… 或保存实验。保存下来的 experiment 会记录完整分析信息,包括手工积分的峰、comments、被排除的 transitions,以及某个 peak 是否被手工接受或拒绝。它使用 .spe 作为扩展名,并且可以在不依赖原始 raw file 或 panel 的情况下重新打开查看数据。
把关键 review 和 refinement 保存进 SPE 后,后续就可以反复回看当时接受某个峰或排除某条 transition 的依据。旧版本实验在升级过程中可能丢失某些 run-level 元数据,例如自定义标签、注释、原始 run 顺序和图形颜色编码,因此 SPE 文件同时也是版本迁移文件。
Next Steps
这里继续进入 Reporting 模块,处理 Post Analysis、Report schema、QC history 和 Appendix 8 report headers,把方法开发结果沉淀为正式验证交付。
继续处理 Post Analysis、Report Perspective、QC 和 Appendix 8 字段训练。
SIS、抗肽抗体免疫富集、复杂基质低丰度读数和 SpectroDive 的承接方式。
Panel、RT calibration 和 method export 对应模块。
查看章节索引、附录和 glossary。